

Description
Galactosialidosis is a condition that affects many areas of the body. The three forms of galactosialidosis are distinguished by the age at which symptoms develop and the pattern of features.
The early infantile form of galactosialidosis is associated with extensive swelling caused by fluid accumulation before birth (hydrops fetalis), a soft out-pouching in the lower abdomen (an inguinal hernia), and an enlarged liver and spleen (hepatosplenomegaly). Additional features of this form include abnormal bone development (dysostosis multiplex) and distinctive facial features that are often described as "coarse." Some infants have an enlarged heart (cardiomegaly), an eye abnormality called a cherry-red spot, and kidney disease that can progress to kidney failure. Infants with this form usually are diagnosed between birth and 3 months of age; they typically live to around 6 months of age.
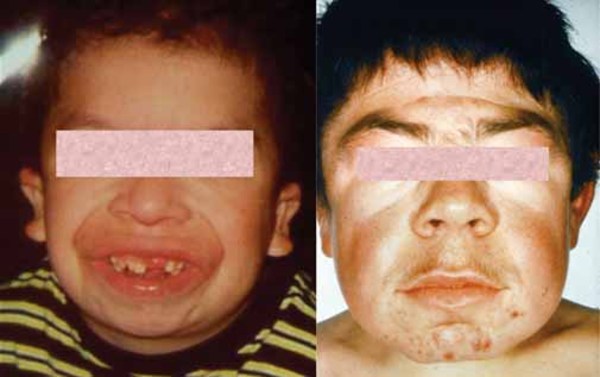

The late infantile form of galactosialidosis shares some features with the early infantile form, although the signs and symptoms are somewhat less severe and begin later in infancy. This form is characterized by short stature, dysostosis multiplex, heart valve problems, hepatosplenomegaly, and "coarse" facial features. Other symptoms seen in some individuals with this type include intellectual disabilities, hearing loss, and a cherry-red spot. Children with this condition typically develop symptoms around 2 years old. The life expectancy of individuals with this type varies depending on the severity of symptoms.
The juvenile/adult form of galactosialidosis has signs and symptoms that are somewhat different from those of the other two types. This form is distinguished by difficulty coordinating movements (ataxia), muscle twitches (myoclonus), seizures, and intellectual disabilities that worsen over time. People with this form typically also have dark red spots on the skin (angiokeratomas), abnormalities in the bones of the spine, "coarse" facial features, a cherry-red spot, vision loss, and hearing loss. The age at which symptoms begin to develop varies widely among affected individuals, but the average age is 16. This form is typically associated with a nearly normal life expectancy.
Frequency
The prevalence of galactosialidosis is unknown; more than 100 cases have been reported. The juvenile/adult form accounts for more than half of the reported cases of galactosialidosis. Most people with this type of the condition are of Japanese descent.
Causes
Variants (also called mutations) in the CTSA gene cause all the forms of galactosialidosis. The CTSA gene provides instructions for making a protein called cathepsin A, which is active in cellular compartments called lysosomes. These compartments contain enzymes that digest and recycle materials when they are no longer needed. Cathepsin A works together with two enzymes, neuraminidase 1 and beta-galactosidase, to form a protein complex. This complex breaks down sugar molecules (oligosaccharides) attached to certain proteins (glycoproteins) or fats (glycolipids). Cathepsin A is also found on the cell surface, where it forms a complex with neuraminidase 1 and a protein called elastin-binding protein. Elastin-binding protein plays a role in the formation of elastic fibers, which are components of the connective tissues that make up the body's supportive framework.

CTSA gene variants interfere with the normal function of cathepsin A. Most of the variants that cause galactosialidosis disrupt the protein structure of cathepsin A, impairing its ability to form complexes with neuraminidase 1, beta-galactosidase, and elastin-binding protein. As a result, these other enzymes are not functional, or they break down prematurely.
Galactosialidosis belongs to a large family of lysosomal storage disorders, each caused by the deficiency of a specific lysosomal enzyme or protein. In people with galactosialidosis, impaired functioning of cathepsin A and other enzymes causes certain substances to accumulate in the lysosomes.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Deficiency of cathepsin A
- Goldberg syndrome
- Lysosomal protective protein deficiency
- Neuraminidase deficiency with beta-galactosidase deficiency
- PPCA deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Groener J, Maaswinkel-Mooy P, Smit V, van der Hoeven M, Bakker J, Campos Y, d'Azzo A. New mutations in two Dutch patients with early infantile galactosialidosis. Mol Genet Metab. 2003 Mar;78(3):222-8. doi: 10.1016/s1096-7192(03)00005-2. Citation on PubMed
- Malvagia S, Morrone A, Caciotti A, Bardelli T, d'Azzo A, Ancora G, Zammarchi E, Donati MA. New mutations in the PPBG gene lead to loss of PPCA protein which affects the level of the beta-galactosidase/neuraminidase complex and the EBP-receptor. Mol Genet Metab. 2004 May;82(1):48-55. doi: 10.1016/j.ymgme.2004.02.007. Citation on PubMed
- Matsumoto N, Gondo K, Kukita J, Higaki K, Paragison RC, Nanba E. A case of galactosialidosis with a homozygous Q49R point mutation. Brain Dev. 2008 Oct;30(9):595-8. doi: 10.1016/j.braindev.2008.01.012. Epub 2008 Apr 18. Citation on PubMed
- Nobeyama Y, Honda M, Niimura M. A case of galactosialidosis. Br J Dermatol. 2003 Aug;149(2):405-9. doi: 10.1046/j.1365-2133.2003.05488.x. Citation on PubMed
- Patel MS, Callahan JW, Zhang S, Chan AK, Unger S, Levin AV, Skomorowski MA, Feigenbaum AS, O'Brien K, Hellmann J, Ryan G, Velsher L, Chitayat D. Early-infantile galactosialidosis: prenatal presentation and postnatal follow-up. Am J Med Genet. 1999 Jul 2;85(1):38-47. Citation on PubMed
- Slama T, Garbade SF, Kolker S, Hoffmann GF, Ries M. Quantitative natural history characterization in a cohort of 142 published cases of patients with galactosialidosis-A cross-sectional study. J Inherit Metab Dis. 2019 Mar;42(2):295-302. doi: 10.1002/jimd.12010. Epub 2019 Jan 28. Citation on PubMed
- Takiguchi K, Itoh K, Shimmoto M, Ozand PT, Doi H, Sakuraba H. Structural and functional study of K453E mutant protective protein/cathepsin A causing the late infantile form of galactosialidosis. J Hum Genet. 2000;45(4):200-6. doi: 10.1007/s100380070027. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.