

Description
Galactosemia is a disorder that affects how the body processes a simple sugar called galactose. A small amount of galactose is present in many foods. It is primarily part of a larger sugar called lactose, which is found in all dairy products and many baby formulas. The signs and symptoms of galactosemia result from an inability to use galactose to produce energy.
Researchers have identified several types of galactosemia. These conditions are each caused by mutations in a particular gene and affect different enzymes involved in breaking down galactose.
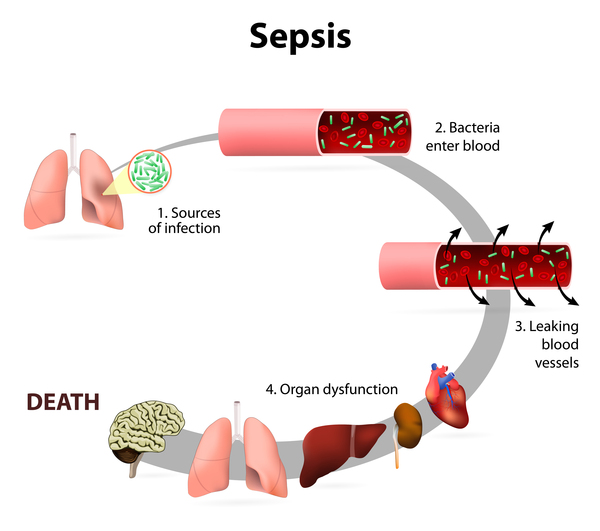
Classic galactosemia, also known as type I, is the most common and most severe form of the condition. If infants with classic galactosemia are not treated promptly with a low-galactose diet, life-threatening complications appear within a few days after birth. Affected infants typically develop feeding difficulties, a lack of energy (lethargy), a failure to gain weight and grow as expected (failure to thrive), yellowing of the skin and whites of the eyes (jaundice), liver damage, and abnormal bleeding. Other serious complications of this condition can include overwhelming bacterial infections (sepsis) and shock. Affected children are also at increased risk of delayed development, clouding of the lens of the eye (cataract), speech difficulties, and intellectual disability. Females with classic galactosemia may develop reproductive problems caused by an early loss of function of the ovaries (premature ovarian insufficiency).
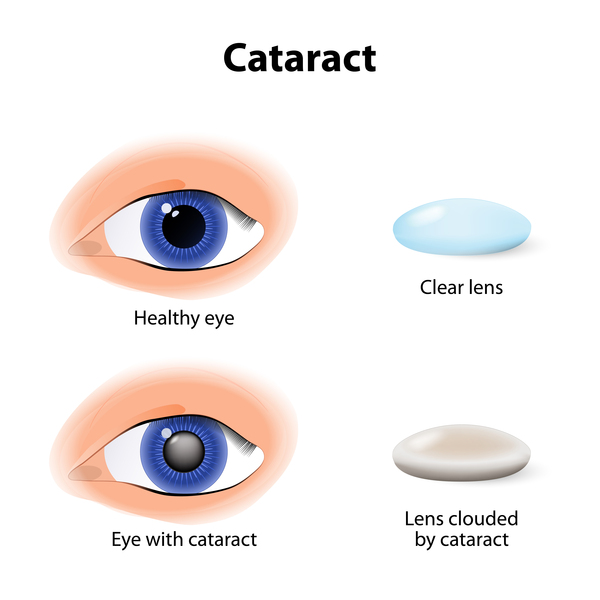
Galactosemia type II (also called galactokinase deficiency) and type III (also called galactose epimerase deficiency) cause different patterns of signs and symptoms. Galactosemia type II causes fewer medical problems than the classic type. Affected infants develop cataracts but otherwise experience few long-term complications. The signs and symptoms of galactosemia type III vary from mild to severe and can include cataracts, delayed growth and development, intellectual disability, liver disease, and kidney problems.
Frequency
Classic galactosemia occurs in 1 in 30,000 to 60,000 newborns. Galactosemia type II and type III are less common; type II probably affects fewer than 1 in 100,000 newborns and type III appears to be very rare.
Causes
Mutations in the GALT, GALK1, and GALE genes cause galactosemia. These genes provide instructions for making enzymes that are essential for processing galactose obtained from the diet. These enzymes break down galactose into another simple sugar, glucose, and other molecules that the body can store or use for energy.
Mutations in the GALT gene cause classic galactosemia (type I). Most of these genetic changes almost completely eliminate the activity of the enzyme produced from the GALT gene, preventing the normal processing of galactose and resulting in the life-threatening signs and symptoms of this disorder. Another GALT gene mutation, known as the Duarte variant, reduces but does not eliminate the activity of the enzyme. People with the Duarte variant tend to have much milder features of galactosemia.
Galactosemia type II results from mutations in the GALK1 gene, while mutations in the GALE gene underlie galactosemia type III. Like the enzyme produced from the GALT gene, the enzymes made from the GALK1 and GALE genes play important roles in processing galactose. A shortage of any of these critical enzymes allows galactose and related compounds to build up to toxic levels in the body. The accumulation of these substances damages tissues and organs, leading to the characteristic features of galactosemia.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Classic galactosemia
- Epimerase deficiency galactosemia
- Galactokinase deficiency disease
- Galactose epimerase deficiency
- Galactose-1-phosphate uridyl-transferase deficiency disease
- GALE deficiency
- GALK deficiency
- GALT deficiency
- UDP-galactose-4-epimerase deficiency disease
- UTP hexose-1-phosphate uridylyltransferase deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Berry GT. Classic Galactosemia and Clinical Variant Galactosemia. 2000 Feb 4 [updated 2021 Mar 11]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1518/ Citation on PubMed
- Berry GT. Galactosemia: when is it a newborn screening emergency? Mol Genet Metab. 2012 May;106(1):7-11. doi: 10.1016/j.ymgme.2012.03.007. Epub 2012 Mar 21. Citation on PubMed
- Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006 Aug;29(4):516-25. doi: 10.1007/s10545-006-0382-0. Epub 2006 Jul 11. Citation on PubMed
- Fridovich-Keil J, Bean L, He M, Schroer R. Epimerase Deficiency Galactosemia. 2011 Jan 25 [updated 2021 Mar 4]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK51671/ Citation on PubMed
- Fridovich-Keil JL, Gambello MJ, Singh RH, Sharer JD. Duarte Variant Galactosemia. 2014 Dec 4 [updated 2020 Jun 25]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK258640/ Citation on PubMed
- Fridovich-Keil JL, Gubbels CS, Spencer JB, Sanders RD, Land JA, Rubio-Gozalbo E. Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis. 2011 Apr;34(2):357-66. doi: 10.1007/s10545-010-9221-4. Epub 2010 Oct 27. Citation on PubMed or Free article on PubMed Central
- Karadag N, Zenciroglu A, Eminoglu FT, Dilli D, Karagol BS, Kundak A, Dursun A, Hakan N, Okumus N. Literature review and outcome of classic galactosemia diagnosed in the neonatal period. Clin Lab. 2013;59(9-10):1139-46. doi: 10.7754/clin.lab.2013.121235. Citation on PubMed
- Openo KK, Schulz JM, Vargas CA, Orton CS, Epstein MP, Schnur RE, Scaglia F, Berry GT, Gottesman GS, Ficicioglu C, Slonim AE, Schroer RJ, Yu C, Rangel VE, Keenan J, Lamance K, Fridovich-Keil JL. Epimerase-deficiency galactosemia is not a binary condition. Am J Hum Genet. 2006 Jan;78(1):89-102. doi: 10.1086/498985. Epub 2005 Nov 14. Citation on PubMed or Free article on PubMed Central
- Timson DJ. The molecular basis of galactosemia - Past, present and future. Gene. 2016 Sep 10;589(2):133-41. doi: 10.1016/j.gene.2015.06.077. Epub 2015 Jul 2. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.