

Description
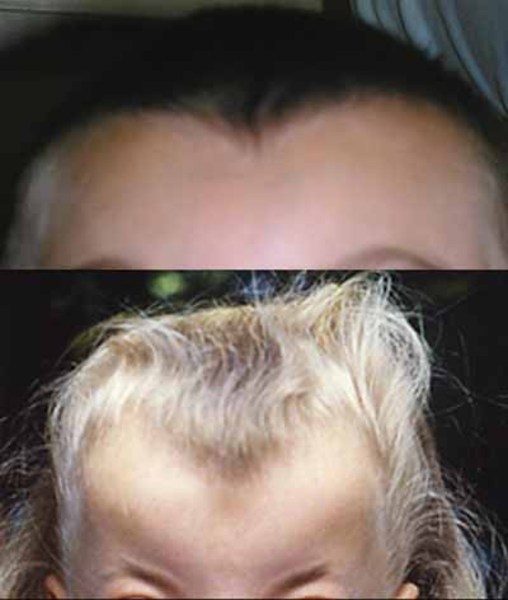
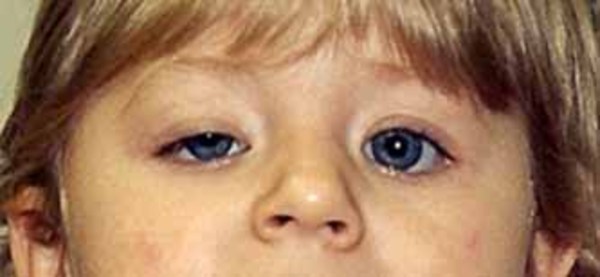
Frontonasal dysplasia is a condition that results from abnormal development of the head and face before birth. People with frontonasal dysplasia have at least two of the following features: widely spaced eyes (ocular hypertelorism); a broad nose; a slit (cleft) in one or both sides of the nose; no nasal tip; a central cleft involving the nose, upper lip, or roof of the mouth (palate); incomplete formation of the front of the skull with skin covering the head where bone should be (anterior cranium bifidum occultum); or a widow's peak hairline.

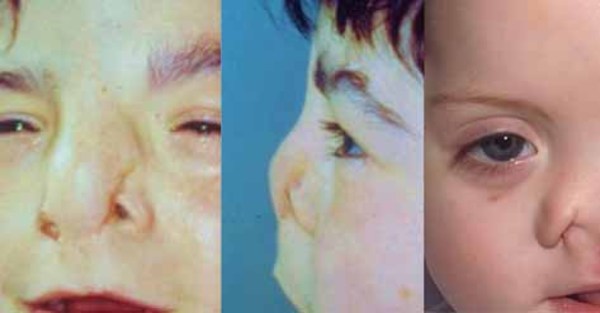

Other features of frontonasal dysplasia can include additional facial malformations, absence or malformation of the tissue that connects the left and right halves of the brain (the corpus callosum), and intellectual disability.

There are at least three types of frontonasal dysplasia that are distinguished by their genetic causes and their signs and symptoms. In addition to the features previously described, each type of frontonasal dysplasia is associated with other distinctive features. Individuals with frontonasal dysplasia type 1 typically have abnormalities of the nose, a long area between the nose and upper lip (philtrum), and droopy upper eyelids (ptosis). Individuals with frontonasal dysplasia type 2 can have hair loss (alopecia) and an enlarged opening in the two bones that make up much of the top and sides of the skull (enlarged parietal foramina). Males with this form of the condition often have genital abnormalities. Features of frontonasal dysplasia type 3 include eyes that are missing (anophthalmia) or very small (microphthalmia) and low-set ears that are rotated backward. Frontonasal dysplasia type 3 is typically associated with the most severe facial abnormalities, but the severity of the condition varies widely, even among individuals with the same type.
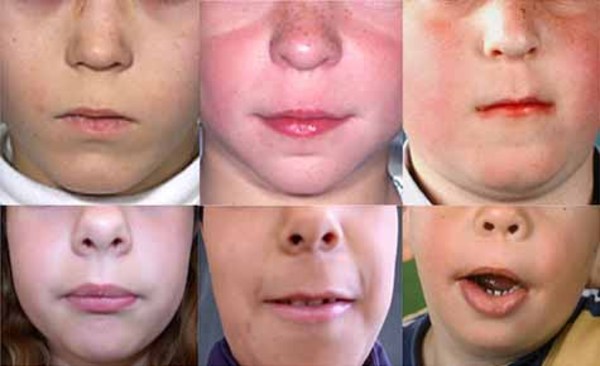
Life expectancy of affected individuals depends on the severity of the malformations and whether or not surgical intervention can improve associated health problems, such as breathing and feeding problems caused by the facial clefts.
Frequency
Frontonasal dysplasia is likely a rare condition; at least 100 cases have been reported in the scientific literature.
Causes
Mutations in the ALX3 gene cause frontonasal dysplasia type 1, ALX4 gene mutations cause type 2, and ALX1 gene mutations cause type 3. These genes provide instructions for making proteins that are necessary for normal development, particularly of the head and face, before birth. The proteins produced from the ALX3, ALX4, and ALX1 genes are transcription factors, which means they attach (bind) to DNA and control the activity of certain genes. Specifically, the proteins control the activity of genes that regulate cell growth and division (proliferation) and movement (migration), ensuring that cells grow and stop growing at specific times and that they are positioned correctly during development. The ALX3 and ALX4 proteins are primarily involved in the development of the nose and surrounding tissues, while the ALX1 protein is involved in development of the eyes, nose, and mouth.
ALX3, ALX4, or ALX1 gene mutations reduce or eliminate function of the respective protein. As a result, the regulation of cell organization during development of the head and face is disrupted, particularly affecting the middle of the face. Abnormal development of the nose, philtrum, and upper lip leads to the facial clefts that characterize this disorder. This abnormal development also interferes with the proper formation of the skull and other facial structures, leading to anterior cranium bifidum occultum, hypertelorism, and other features of frontonasal dysplasia.
Inheritance
When frontonasal dysplasia is caused by mutations in the ALX1 or ALX3 gene, it is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

When ALX4 gene mutations cause frontonasal dysplasia, the condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


Other Names for This Condition
- FND
- FNM
- Frontonasal dysplasia sequence
- Frontonasal malformation
- Frontorhiny
- Median facial cleft syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Allam KA, Wan DC, Kawamoto HK, Bradley JP, Sedano HO, Saied S. The spectrum of median craniofacial dysplasia. Plast Reconstr Surg. 2011 Feb;127(2):812-821. doi: 10.1097/PRS.0b013e318200aa08. Citation on PubMed
- Giffoni SD, Goncalves VM, Zanardi VA, Lopes VL. Angular analysis of corpus callosum in 18 patients with frontonasal dysplasia. Arq Neuropsiquiatr. 2004 Jun;62(2A):195-8. doi: 10.1590/s0004-282x2004000200001. Epub 2004 Jun 23. Citation on PubMed
- Kayserili H, Altunoglu U, Ozgur H, Basaran S, Uyguner ZO. Mild nasal malformations and parietal foramina caused by homozygous ALX4 mutations. Am J Med Genet A. 2012 Jan;158A(1):236-44. doi: 10.1002/ajmg.a.34390. Epub 2011 Dec 2. Citation on PubMed
- Kayserili H, Uz E, Niessen C, Vargel I, Alanay Y, Tuncbilek G, Yigit G, Uyguner O, Candan S, Okur H, Kaygin S, Balci S, Mavili E, Alikasifoglu M, Haase I, Wollnik B, Akarsu NA. ALX4 dysfunction disrupts craniofacial and epidermal development. Hum Mol Genet. 2009 Nov 15;18(22):4357-66. doi: 10.1093/hmg/ddp391. Epub 2009 Aug 19. Citation on PubMed
- Pham NS, Rafii A, Liu J, Boyadjiev SA, Tollefson TT. Clinical and genetic characterization of frontorhiny: report of 3 novel cases and discussion of the surgical management. Arch Facial Plast Surg. 2011 Nov-Dec;13(6):415-20. doi: 10.1001/archfacial.2011.684. Citation on PubMed
- Roarty JD, Pron GE, Siegel-Bartelt J, Posnick JC, Buncic JR. Ocular manifestations of frontonasal dysplasia. Plast Reconstr Surg. 1994 Jan;93(1):25-30. doi: 10.1097/00006534-199401000-00004. Citation on PubMed
- Twigg SR, Versnel SL, Nurnberg G, Lees MM, Bhat M, Hammond P, Hennekam RC, Hoogeboom AJ, Hurst JA, Johnson D, Robinson AA, Scambler PJ, Gerrelli D, Nurnberg P, Mathijssen IM, Wilkie AO. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am J Hum Genet. 2009 May;84(5):698-705. doi: 10.1016/j.ajhg.2009.04.009. Epub 2009 Apr 30. Citation on PubMed or Free article on PubMed Central
- Uz E, Alanay Y, Aktas D, Vargel I, Gucer S, Tuncbilek G, von Eggeling F, Yilmaz E, Deren O, Posorski N, Ozdag H, Liehr T, Balci S, Alikasifoglu M, Wollnik B, Akarsu NA. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am J Hum Genet. 2010 May 14;86(5):789-96. doi: 10.1016/j.ajhg.2010.04.002. Epub 2010 May 6. Citation on PubMed or Free article on PubMed Central
- Wu E, Vargevik K, Slavotinek AM. Subtypes of frontonasal dysplasia are useful in determining clinical prognosis. Am J Med Genet A. 2007 Dec 15;143A(24):3069-78. doi: 10.1002/ajmg.a.31963. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.