

Description
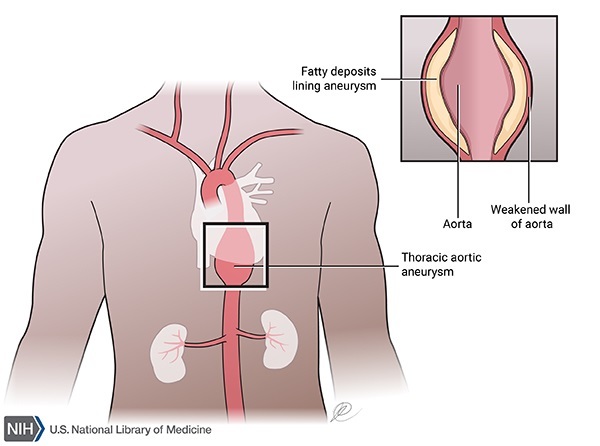
Familial thoracic aortic aneurysm and dissection (familial TAAD) involves problems with the aorta, which is the large blood vessel that distributes blood from the heart to the rest of the body. Familial TAAD affects the upper part of the aorta, near the heart. This part of the aorta is called the thoracic aorta because it is located in the chest (thorax). Other vessels that carry blood from the heart to the rest of the body (arteries) can also be affected.

In familial TAAD, the aorta can become weakened and stretched (aortic dilatation), which can lead to a bulge in the blood vessel wall (an aneurysm). Aortic dilatation may also lead to a sudden tearing of the layers in the aorta wall (aortic dissection), allowing blood to flow abnormally between the layers. These aortic abnormalities are potentially life-threatening because they can decrease blood flow to other parts of the body such as the brain or other vital organs, or cause the aorta to break open (rupture).
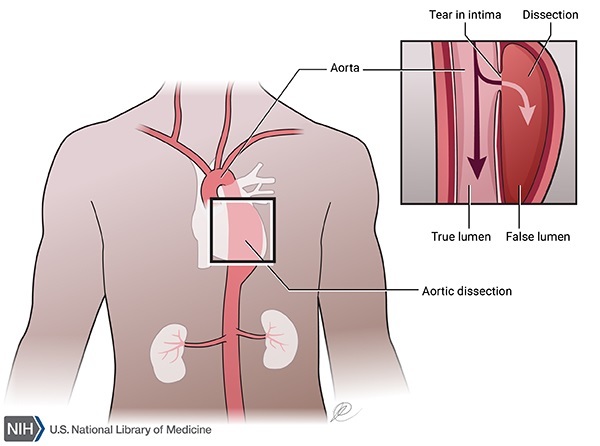
The occurrence and timing of these aortic abnormalities vary, even within the same affected family. They can begin in childhood or not occur until late in life. Aortic dilatation is generally the first feature of familial TAAD to develop, although in some affected individuals dissection occurs with little or no aortic dilatation.
Aortic aneurysms usually have no symptoms. However, depending on the size, growth rate, and location of these abnormalities, they can cause pain in the jaw, neck, chest, or back; swelling in the arms, neck, or head; difficult or painful swallowing; hoarseness; shortness of breath; wheezing; a chronic cough; or coughing up blood. Aortic dissections usually cause severe, sudden chest or back pain, and may also result in unusually pale skin (pallor), a very faint pulse, numbness or tingling (paresthesias) in one or more limbs, or paralysis.
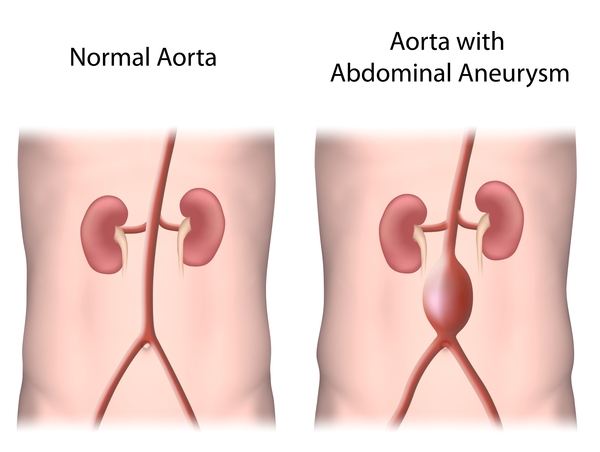
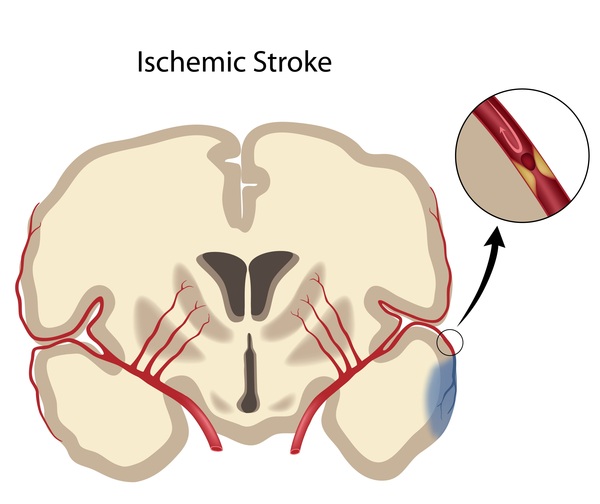
Familial TAAD may not be associated with other signs and symptoms. However, some individuals in affected families show mild features of related conditions called Marfan syndrome or Loeys-Dietz syndrome. These features include tall stature, stretch marks on the skin, an unusually large range of joint movement (joint hypermobility), and either a sunken or protruding chest. Occasionally, people with familial TAAD develop aneurysms in the brain or in the section of the aorta located in the abdomen (abdominal aorta). Some people with familial TAAD have heart abnormalities that are present from birth (congenital). Affected individuals may also have a soft out-pouching in the lower abdomen (inguinal hernia), an abnormal curvature of the spine (scoliosis), or a purplish skin discoloration (livedo reticularis) caused by abnormalities in the tiny blood vessels of the skin (dermal capillaries). However, these conditions are also common in the general population. Depending on the genetic cause of familial TAAD in particular families, they may have an increased risk of developing blockages in smaller arteries, which can lead to heart attack and stroke.

Frequency
Familial TAAD is believed to account for at least 20 percent of thoracic aortic aneurysms and dissections. In the remainder of cases, the abnormalities are thought to be caused by factors that are not inherited, such as damage to the walls of the aorta from aging, tobacco use, injury, or disease.
While aortic aneurysms are common worldwide, it is difficult to determine their exact prevalence because they usually cause no symptoms unless they rupture. Ruptured aortic aneurysms and dissections are estimated to cause almost 30,000 deaths in the United States each year.
Causes
Mutations in any of several genes are associated with familial TAAD. Mutations in the ACTA2 gene have been identified in 14 to 20 percent of people with this disorder, and TGFBR2 gene mutations have been found in 2.5 percent of affected individuals. Mutations in several other genes account for smaller percentages of cases.
The ACTA2 gene provides instructions for making a protein called smooth muscle alpha (α)-2 actin, which is found in vascular smooth muscle cells. Layers of these cells are found in the walls of the aorta and other arteries. Within vascular smooth muscle cells, smooth muscle α-2 actin forms the core of structures called sarcomeres, which are necessary for muscles to contract. This ability to contract allows the arteries to maintain their shape instead of stretching out as blood is pumped through them.
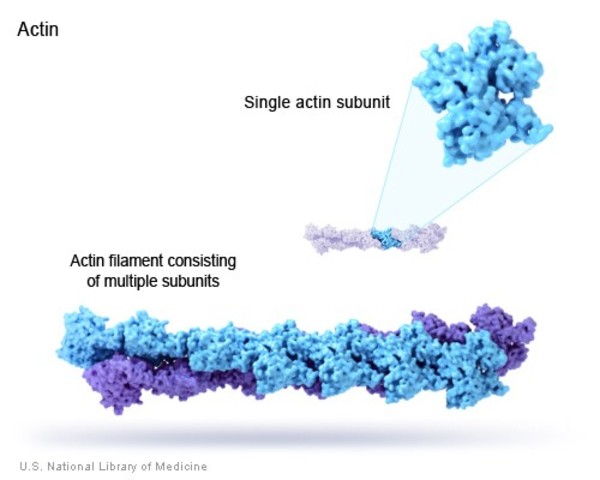
ACTA2 gene mutations that are associated with familial TAAD change single protein building blocks (amino acids) in the smooth muscle α-2 actin protein. These changes likely affect the way the protein functions in smooth muscle contraction, interfering with the sarcomeres' ability to prevent the arteries from stretching. The aorta, where the force of blood pumped directly from the heart is most intense, is particularly vulnerable to this stretching. Abnormal stretching of the aorta results in the aortic dilatation, aneurysms, and dissections that characterize familial TAAD.

TGFBR2 gene mutations are also associated with familial TAAD. The TGFBR2 gene provides instructions for making a protein called transforming growth factor-beta (TGF-β) receptor type 2. This receptor transmits signals from the cell surface into the cell through a process called signal transduction. Through this type of signaling, the environment outside the cell affects activities inside the cell. In particular, the TGF-β receptor type 2 protein helps control the growth and division (proliferation) of cells and the process by which cells mature to carry out specific functions (differentiation). It is also involved in the formation of the extracellular matrix, an intricate lattice of proteins and other molecules that forms in the spaces between cells.
TGFBR2 gene mutations alter the receptor's structure, which disturbs signal transduction. The disturbed signaling can impair cell growth and development. It is not known how these changes result in the specific aortic abnormalities associated with familial TAAD.
Mutations in other genes, some of which have not been identified, are also associated with familial TAAD.
Inheritance
Familial TAAD is inherited in an autosomal dominant pattern, which means one copy of an altered gene in each cell can be sufficient to cause the condition. In most cases, an affected person has one affected parent. However, some people who inherit an altered gene never develop the aortic abnormalities associated with the condition; this situation is known as reduced penetrance.

Other Names for This Condition
- Annuloaortic ectasia
- Congenital aneurysm of ascending aorta
- FAA
- Familial aortic aneurysm
- Familial aortic dissection
- Familial TAAD
- Familial thoracic aortic aneurysm
- FTAAD
- TAA
- TAAD
- Thoracic aortic aneurysm
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Brautbar A, LeMaire SA, Franco LM, Coselli JS, Milewicz DM, Belmont JW. FBN1 mutations in patients with descending thoracic aortic dissections. Am J Med Genet A. 2010 Feb;152A(2):413-6. doi: 10.1002/ajmg.a.32856. Citation on PubMed or Free article on PubMed Central
- El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 2009 Dec;6(12):771-86. doi: 10.1038/nrcardio.2009.191. Epub 2009 Nov 3. Citation on PubMed
- Elefteriades JA, Pomianowski P. Practical genetics of thoracic aortic aneurysm. Prog Cardiovasc Dis. 2013 Jul-Aug;56(1):57-67. doi: 10.1016/j.pcad.2013.06.002. Citation on PubMed
- Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, Grabenwoger M, Haverich A, Iung B, Manolis AJ, Meijboom F, Nienaber CA, Roffi M, Rousseau H, Sechtem U, Sirnes PA, Allmen RS, Vrints CJ; ESC Committee for Practice Guidelines. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014 Nov 1;35(41):2873-926. doi: 10.1093/eurheartj/ehu281. Epub 2014 Aug 29. No abstract available. Erratum In: Eur Heart J. 2015 Nov 1;36(41):2779. Citation on PubMed
- Grond-Ginsbach C, Pjontek R, Aksay SS, Hyhlik-Durr A, Bockler D, Gross-Weissmann ML. Spontaneous arterial dissection: phenotype and molecular pathogenesis. Cell Mol Life Sci. 2010 Jun;67(11):1799-815. doi: 10.1007/s00018-010-0276-z. Epub 2010 Feb 14. Citation on PubMed
- Guo DC, Regalado E, Casteel DE, Santos-Cortez RL, Gong L, Kim JJ, Dyack S, Horne SG, Chang G, Jondeau G, Boileau C, Coselli JS, Li Z, Leal SM, Shendure J, Rieder MJ, Bamshad MJ, Nickerson DA; GenTAC Registry Consortium; National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project; Kim C, Milewicz DM. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013 Aug 8;93(2):398-404. doi: 10.1016/j.ajhg.2013.06.019. Epub 2013 Aug 1. Citation on PubMed or Free article on PubMed Central
- Jondeau G, Boileau C. Genetics of thoracic aortic aneurysms. Curr Atheroscler Rep. 2012 Jun;14(3):219-26. doi: 10.1007/s11883-012-0241-4. Citation on PubMed
- Milewicz DM, Carlson AA, Regalado ES. Genetic testing in aortic aneurysm disease: PRO. Cardiol Clin. 2010 May;28(2):191-7. doi: 10.1016/j.ccl.2010.01.017. Citation on PubMed or Free article on PubMed Central
- Milewicz DM, Cecchi AC. Heritable Thoracic Aortic Disease Overview. 2003 Feb 13 [updated 2023 May 4]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1120/ Citation on PubMed
- Morisaki H, Akutsu K, Ogino H, Kondo N, Yamanaka I, Tsutsumi Y, Yoshimuta T, Okajima T, Matsuda H, Minatoya K, Sasaki H, Tanaka H, Ishibashi-Ueda H, Morisaki T. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum Mutat. 2009 Oct;30(10):1406-11. doi: 10.1002/humu.21081. Citation on PubMed
- Pyeritz RE. Heritable thoracic aortic disorders. Curr Opin Cardiol. 2014 Jan;29(1):97-102. doi: 10.1097/HCO.0000000000000023. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.