

Description
Familial dilated cardiomyopathy is a genetic form of heart disease. It occurs when heart (cardiac) muscle becomes thin and weakened in at least one chamber of the heart, causing the open area of the chamber to become enlarged (dilated). As a result, the heart is unable to pump blood as efficiently as usual. To compensate, the heart attempts to increase the amount of blood being pumped through the heart, leading to further thinning and weakening of the cardiac muscle. Over time, this condition results in heart failure.
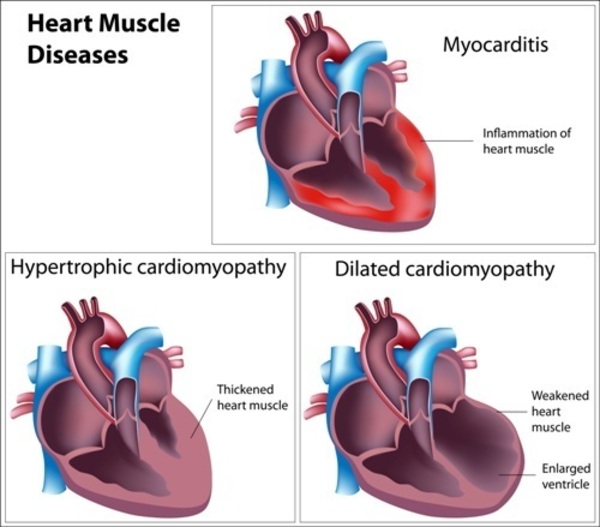
It usually takes many years for symptoms of familial dilated cardiomyopathy to cause health problems. They typically begin in mid-adulthood, but can occur at any time from infancy to late adulthood. Signs and symptoms of familial dilated cardiomyopathy can include an irregular heartbeat (arrhythmia), shortness of breath (dyspnea), extreme tiredness (fatigue), fainting episodes (syncope), and swelling of the legs and feet. In some cases, the first sign of the disorder is sudden cardiac death. The severity of the condition varies among affected individuals, even in members of the same family.
Frequency
It is estimated that 750,000 people in the United States have dilated cardiomyopathy; roughly half of these cases are familial.
Causes
Mutations in more than 30 genes have been found to cause familial dilated cardiomyopathy. These genes provide instructions for making proteins that are found in cardiac muscle cells called cardiomyocytes.
Many of these proteins play important roles in the contraction of the cardiac muscle through their association with cell structures called sarcomeres. Sarcomeres are the basic units of muscle contraction; they are made of proteins that generate the mechanical force needed for muscles to contract. Many other proteins associated with familial dilated cardiomyopathy make up the structural framework (the cytoskeleton) of cardiomyocytes. The remaining proteins play various roles within cardiomyocytes to ensure their proper functioning.
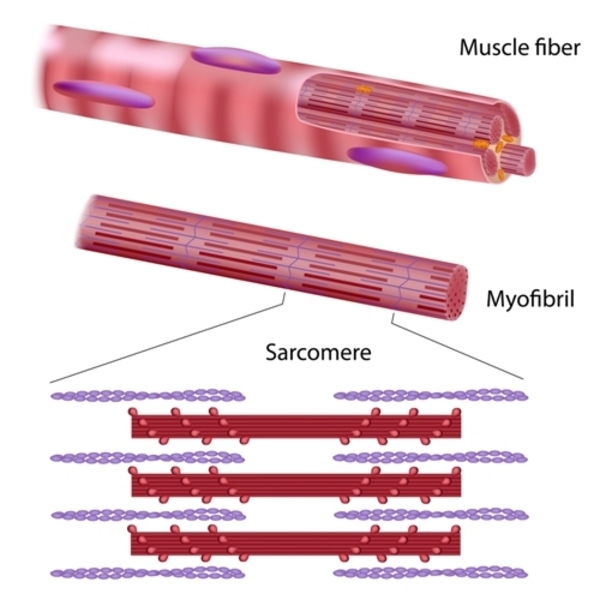
Mutations in one gene, TTN, account for approximately 20 percent of cases of familial dilated cardiomyopathy. The TTN gene provides instructions for making a protein called titin, which is found in the sarcomeres of many types of muscle cells, including cardiomyocytes. Titin provides structure, flexibility, and stability to sarcomeres. Titin also plays a role in chemical signaling and in assembling new sarcomeres. The TTN gene mutations that cause familial dilated cardiomyopathy result in the production of an abnormally short titin protein. It is unclear how the altered protein causes familial dilated cardiomyopathy, but it is likely that it impairs sarcomere function and disrupts chemical signaling.
It is unclear how mutations in the other genes cause familial dilated cardiomyopathy. It is likely that the changes impair cardiomyocyte function and reduce the ability of these cells to contract, weakening and thinning cardiac muscle.
People with familial dilated cardiomyopathy often do not have an identified mutation in any of the known associated genes. The cause of the condition in these individuals is unknown.
Familial dilated cardiomyopathy is described as nonsyndromic or isolated because it typically affects only the heart. However, dilated cardiomyopathy can also occur as part of syndromes that affect other organs and tissues in the body. These forms of the condition are described as syndromic and are caused by mutations in other genes. Additionally, there are many nongenetic causes of dilated cardiomyopathy, including viral infection and chronic alcohol abuse.
Inheritance
Familial dilated cardiomyopathy has different inheritance patterns depending on the gene involved.
In 80 to 90 percent of cases, familial dilated cardiomyopathy is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person inherits the mutation from one affected parent. However, some people who inherit the altered gene never develop features of familial dilated cardiomyopathy. (This situation is known as reduced penetrance.) Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


In rare instances, this condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

In other rare cases, this condition is inherited in an X-linked pattern. In these cases, the gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell increases the risk of developing heart disease, but females with such a mutation may not develop familial dilated cardiomyopathy. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes familial dilated cardiomyopathy. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Other Names for This Condition
- Congestive cardiomyopathy
- Familial idiopathic cardiomyopathy
- FDC
- Primary familial dilated cardiomyopathy
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- CARDIOMYOPATHY, DILATED, 1A; CMD1A
- CARDIOMYOPATHY, DILATED, 1C, WITH OR WITHOUT LEFT VENTRICULAR NONCOMPACTION; CMD1C
- CARDIOMYOPATHY, DILATED, 1D; CMD1D
- CARDIOMYOPATHY, DILATED, 1B; CMD1B
- CARDIOMYOPATHY, DILATED, 3B; CMD3B
- CARDIOMYOPATHY, DILATED, 1E; CMD1E
- CARDIOMYOPATHY, DILATED, 1I; CMD1I
- CARDIOMYOPATHY, DILATED, 1J; CMD1J
- CARDIOMYOPATHY, DILATED, 1G; CMD1G
- CARDIOMYOPATHY, DILATED, 1H; CMD1H
- CARDIOMYOPATHY, DILATED, 1O; CMD1O
- CARDIOMYOPATHY, DILATED, 1L; CMD1L
- CARDIOMYOPATHY, DILATED, 1K; CMD1K
- CARDIOMYOPATHY, DILATED, 1M; CMD1M
- CARDIOMYOPATHY, FAMILIAL HYPERTROPHIC, 25; CMH25
- CARDIOMYOPATHY, DILATED, 1P; CMD1P
- CARDIOMYOPATHY, DILATED, 1Q; CMD1Q
- CARDIOMYOPATHY, DILATED, 1W; CMD1W
- CARDIOMYOPATHY, DILATED, 1Y; CMD1Y
- CARDIOMYOPATHY, DILATED, 1Z; CMD1Z
- CARDIOMYOPATHY, DILATED, 2A; CMD2A
- CARDIOMYOPATHY, DILATED, 1AA, WITH OR WITHOUT LEFT VENTRICULAR NONCOMPACTION; CMD1AA
- LEFT VENTRICULAR NONCOMPACTION 10; LVNC10
- CARDIOMYOPATHY, DILATED, 1DD; CMD1DD
- CARDIOMYOPATHY, DILATED, 1KK; CMD1KK
- CARDIOMYOPATHY, DILATED, 1U; CMD1U
- CARDIOMYOPATHY, DILATED, 1V; CMD1V
- CARDIOMYOPATHY, DILATED, 1EE; CMD1EE
- CARDIOMYOPATHY, DILATED, 1R; CMD1R
- CARDIOMYOPATHY, DILATED, 1S; CMD1S
- CARDIOMYOPATHY, DILATED, 1JJ; CMD1JJ
- CARDIOMYOPATHY, DILATED, 1HH; CMD1HH
- CARDIOMYOPATHY, DILATED, 2B; CMD2B
- CARDIOMYOPATHY, DILATED, 1II; CMD1II
- CARDIOMYOPATHY, DILATED, 1BB; CMD1BB
Scientific Articles on PubMed
References
- Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011 Aug;8(8):1308-39. doi: 10.1016/j.hrthm.2011.05.020. No abstract available. Citation on PubMed
- Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2010 May;25(3):198-204. doi: 10.1097/HCO.0b013e328337ba52. Citation on PubMed or Free article on PubMed Central
- Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012 Feb 16;366(7):619-28. doi: 10.1056/NEJMoa1110186. Citation on PubMed or Free article on PubMed Central
- Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013 Sep;10(9):531-47. doi: 10.1038/nrcardio.2013.105. Epub 2013 Jul 30. Citation on PubMed
- Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010 Nov;12(11):655-67. doi: 10.1097/GIM.0b013e3181f2481f. Citation on PubMed or Free article on PubMed Central
- Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011 Apr 19;57(16):1641-9. doi: 10.1016/j.jacc.2011.01.015. Citation on PubMed or Free article on PubMed Central
- Morales A, Hershberger RE. Genetic evaluation of dilated cardiomyopathy. Curr Cardiol Rep. 2013 Jul;15(7):375. doi: 10.1007/s11886-013-0375-1. Citation on PubMed
- Posafalvi A, Herkert JC, Sinke RJ, van den Berg MP, Mogensen J, Jongbloed JD, van Tintelen JP. Clinical utility gene card for: dilated cardiomyopathy (CMD). Eur J Hum Genet. 2013 Oct;21(10). doi: 10.1038/ejhg.2012.276. Epub 2012 Dec 19. No abstract available. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.