

Description
Esophageal atresia/tracheoesophageal fistula (EA/TEF) is a condition resulting from abnormal development before birth of the tube that carries food from the mouth to the stomach (the esophagus). During early development, the esophagus and windpipe (trachea) begin as a single tube that normally divides into the two adjacent passages between four and eight weeks after conception. If this separation does not occur properly, EA/TEF is the result.
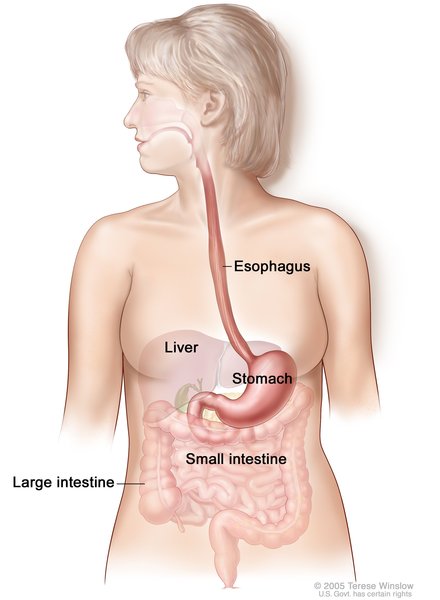
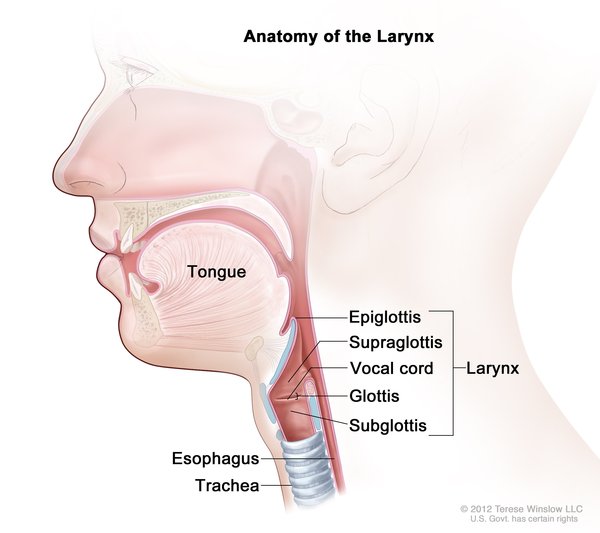
In esophageal atresia (EA), the upper esophagus does not connect (atresia) to the lower esophagus and stomach. Almost 90 percent of babies born with esophageal atresia also have a tracheoesophageal fistula (TEF), in which the esophagus and the trachea are abnormally connected, allowing fluids from the esophagus to get into the airways and interfere with breathing. A small number of infants have only one of these abnormalities.
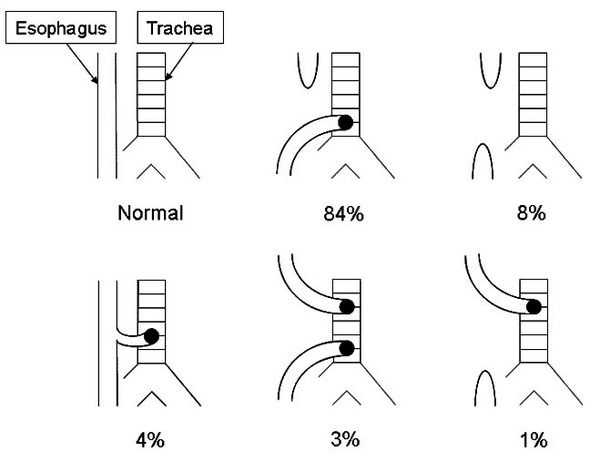
There are several types of EA/TEF, classified by the location of the malformation and the structures that are affected. In more than 80 percent of cases, the lower section of the malformed esophagus is connected to the trachea (EA with a distal TEF). Other possible configurations include having the upper section of the malformed esophagus connected to the trachea (EA with a proximal TEF), connections to the trachea from both the upper and lower sections of the malformed esophagus (EA with proximal and distal TEF), an esophagus that is malformed but does not connect to the trachea (isolated EA), and a connection to the trachea from an otherwise normal esophagus (H-type TEF with no EA).
While EA/TEF arises during fetal development, it generally becomes apparent shortly after birth. Saliva, liquids fed to the infant, or digestive fluids may enter the windpipe through the tracheoesophageal fistula, leading to coughing, respiratory distress, and a bluish appearance of the skin or lips (cyanosis). Esophageal atresia blocks liquids fed to the infant from entering the stomach, so they are spit back up, sometimes along with fluids from the respiratory tract. EA/TEF is a life-threatening condition; affected babies generally require surgery to correct the malformation in order to allow feeding and prevent lung damage from repeated exposure to esophageal fluids.
EA/TEF occurs alone (isolated EA/TEF) in about 40 percent of affected individuals. In other cases it occurs with other birth defects or as part of a genetic syndrome (non-isolated or syndromic EA/TEF).
Frequency
EA/TEF occurs in 1 in 3,000 to 5,000 newborns.
Causes
Isolated EA/TEF is considered to be a multifactorial condition, which means that multiple gene variations and environmental factors likely contribute to its occurrence. In most cases of isolated EA/TEF, no specific genetic changes or environmental factors have been conclusively determined to be the cause.
Non-isolated or syndromic forms of EA/TEF can be caused by changes in single genes or in chromosomes, or they can be multifactorial. For example, approximately 10 percent of people with CHARGE syndrome, which is usually caused by mutations in the CHD7 gene, have EA/TEF. About 25 percent of individuals with the chromosomal abnormality trisomy 18 are born with EA/TEF. EA/TEF also occurs in VACTERL association, a multifactorial condition. VACTERL is an acronym that stands for vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula, renal anomalies, and limb abnormalities. People diagnosed with VACTERL association typically have at least three of these features; between 50 and 80 percent have a tracheoesophageal fistula.
Inheritance
When EA/TEF occurs as a feature of a genetic syndrome or chromosomal abnormality, it may cluster in families according to the inheritance pattern for that condition. Often EA/TEF is not inherited, and there is only one affected individual in a family.
Other Names for This Condition
- EA/TEF
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Achildi O, Grewal H. Congenital anomalies of the esophagus. Otolaryngol Clin North Am. 2007 Feb;40(1):219-44, viii. doi: 10.1016/j.otc.2006.10.010. Citation on PubMed
- Bednarczyk D, Sasiadek MM, Smigiel R. Chromosome aberrations and gene mutations in patients with esophageal atresia. J Pediatr Gastroenterol Nutr. 2013 Dec;57(6):688-93. doi: 10.1097/MPG.0b013e3182a373dc. Citation on PubMed
- Brosens E, Ploeg M, van Bever Y, Koopmans AE, IJsselstijn H, Rottier RJ, Wijnen R, Tibboel D, de Klein A. Clinical and etiological heterogeneity in patients with tracheo-esophageal malformations and associated anomalies. Eur J Med Genet. 2014 Aug;57(8):440-52. doi: 10.1016/j.ejmg.2014.05.009. Epub 2014 Jun 13. Citation on PubMed
- Brunner HG, van Bokhoven H. Genetic players in esophageal atresia and tracheoesophageal fistula. Curr Opin Genet Dev. 2005 Jun;15(3):341-7. doi: 10.1016/j.gde.2005.04.010. Citation on PubMed
- de Jong EM, Felix JF, de Klein A, Tibboel D. Etiology of esophageal atresia and tracheoesophageal fistula: "mind the gap". Curr Gastroenterol Rep. 2010 Jun;12(3):215-22. doi: 10.1007/s11894-010-0108-1. Citation on PubMed or Free article on PubMed Central
- El-Gohary Y, Gittes GK, Tovar JA. Congenital anomalies of the esophagus. Semin Pediatr Surg. 2010 Aug;19(3):186-93. doi: 10.1053/j.sempedsurg.2010.03.009. Citation on PubMed
- Felix JF, de Jong EM, Torfs CP, de Klein A, Rottier RJ, Tibboel D. Genetic and environmental factors in the etiology of esophageal atresia and/or tracheoesophageal fistula: an overview of the current concepts. Birth Defects Res A Clin Mol Teratol. 2009 Sep;85(9):747-54. doi: 10.1002/bdra.20592. Citation on PubMed
- Felix JF, Tibboel D, de Klein A. Chromosomal anomalies in the aetiology of oesophageal atresia and tracheo-oesophageal fistula. Eur J Med Genet. 2007 May-Jun;50(3):163-75. doi: 10.1016/j.ejmg.2006.12.004. Epub 2007 Jan 21. Citation on PubMed
- Genevieve D, de Pontual L, Amiel J, Sarnacki S, Lyonnet S. An overview of isolated and syndromic oesophageal atresia. Clin Genet. 2007 May;71(5):392-9. doi: 10.1111/j.1399-0004.2007.00798.x. Citation on PubMed
- Scott DA. Esophageal Atresia / Tracheoesophageal Fistula Overview - RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. 2009 Mar 12 [updated 2018 Sep 20]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK5192/ Citation on PubMed
- Spitz L. Oesophageal atresia. Orphanet J Rare Dis. 2007 May 11;2:24. doi: 10.1186/1750-1172-2-24. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.