

Description
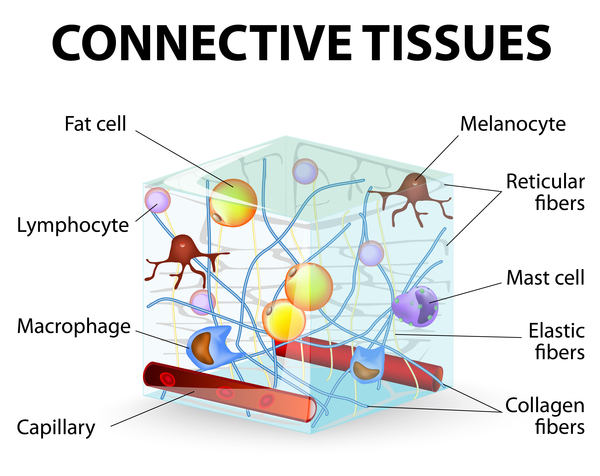
Ehlers-Danlos syndrome is a group of disorders that affect connective tissues supporting the skin, bones, blood vessels, and many other organs and tissues. Defects in connective tissues cause the signs and symptoms of these conditions, which range from mildly loose joints to life-threatening complications.
The various forms of Ehlers-Danlos syndrome have been classified in several different ways. Originally, 11 forms of Ehlers-Danlos syndrome were named using Roman numerals to indicate the types (type I, type II, and so on). In 1997, researchers proposed a simpler classification (the Villefranche nomenclature) that reduced the number of types to six and gave them descriptive names based on their major features. In 2017, the classification was updated to include rare forms of Ehlers-Danlos syndrome that were identified more recently. The 2017 classification describes 13 types of Ehlers-Danlos syndrome.
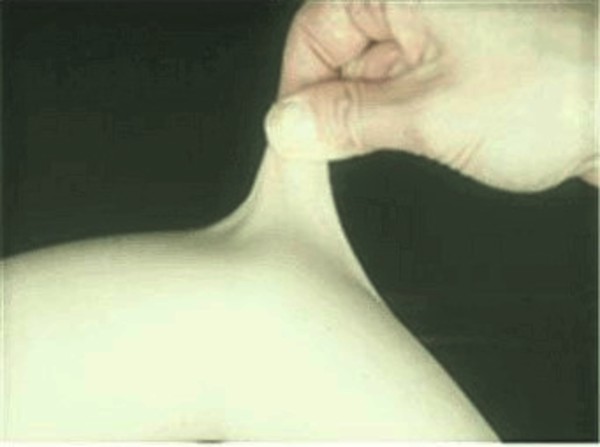
An unusually large range of joint movement (hypermobility) occurs in most forms of Ehlers-Danlos syndrome, and it is a hallmark feature of the hypermobile type. Infants and children with hypermobility often have weak muscle tone (hypotonia), which can delay the development of motor skills such as sitting, standing, and walking. The loose joints are unstable and prone to dislocation and chronic pain. In the arthrochalasia type of Ehlers-Danlos syndrome, infants have hypermobility and dislocations of both hips at birth.
Many people with the Ehlers-Danlos syndromes have soft, velvety skin that is highly stretchy (elastic) and fragile. Affected individuals tend to bruise easily, and some types of the condition also cause abnormal scarring. People with the classical form of Ehlers-Danlos syndrome experience wounds that split open with little bleeding and leave scars that widen over time to create characteristic "cigarette paper" scars. The dermatosparaxis type of the disorder is characterized by loose skin that sags and wrinkles, and extra (redundant) folds of skin may be present.
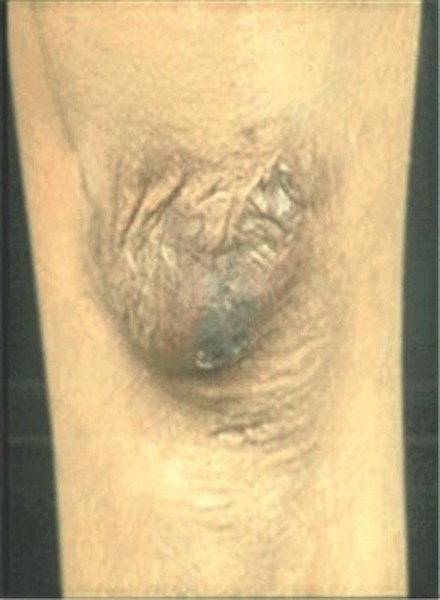
Bleeding problems are common in the vascular type of Ehlers-Danlos syndrome and are caused by unpredictable tearing (rupture) of blood vessels and organs. These complications can lead to easy bruising, internal bleeding, a hole in the wall of the intestine (intestinal perforation), or stroke. During pregnancy, women with vascular Ehlers-Danlos syndrome may experience rupture of the uterus. Additional forms of Ehlers-Danlos syndrome that involve rupture of the blood vessels include the kyphoscoliotic, classical, and classical-like types.
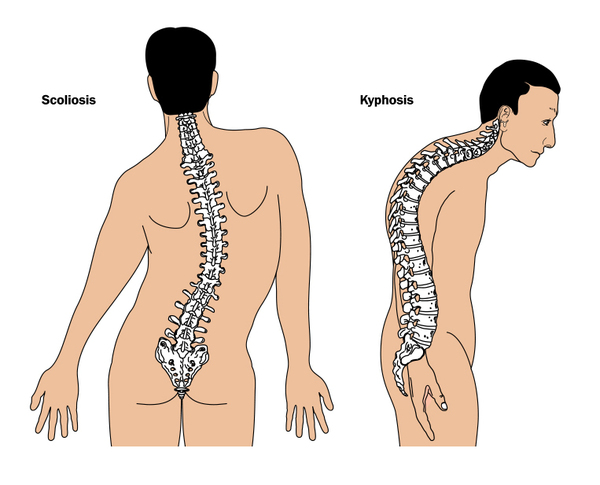
Other types of Ehlers-Danlos syndrome have additional signs and symptoms. The cardiac-valvular type causes severe problems with the valves that control the movement of blood through the heart. People with the kyphoscoliotic type experience severe curvature of the spine that worsens over time and can interfere with breathing by restricting lung expansion. A type of Ehlers-Danlos syndrome called brittle cornea syndrome is characterized by thinness of the clear covering of the eye (the cornea) and other eye abnormalities. The spondylodysplastic type features short stature and skeletal abnormalities such as abnormally curved (bowed) limbs. Abnormalities of muscles, including hypotonia and permanently bent joints (contractures), are among the characteristic signs of the musculocontractural and myopathic forms of Ehlers-Danlos syndrome. The periodontal type causes abnormalities of the teeth and gums.
Frequency
The combined prevalence of all types of Ehlers-Danlos syndrome appears to be at least 1 in 5,000 individuals worldwide. The hypermobile and classical forms are most common; the hypermobile type may affect as many as 1 in 5,000 to 20,000 people, while the classical type probably occurs in 1 in 20,000 to 40,000 people. Other forms of Ehlers-Danlos syndrome are rare, often with only a few cases or affected families described in the medical literature.
Causes
Variants (also known as mutations) in at least 20 genes have been found to cause the Ehlers-Danlos syndromes. Variants in the COL5A1 or COL5A2 gene, or rarely in the COL1A1 gene, can cause the classical type. Variants in the TNXB gene cause the classical-like type and have been reported in a very small percentage of cases of the hypermobile type (although in most people with this type, the cause is unknown). The cardiac-valvular type and some cases of the arthrochalasia type are caused by COL1A2 gene variants; variants in the COL1A1 gene have also been found in people with the arthrochalasia type. Most cases of the vascular type result from variants in the COL3A1 gene, although rarely this type is caused by certain COL1A1 gene variants. The dermatosparaxis type is caused by variants in the ADAMTS2 gene. PLOD1 or FKBP14 gene variants result in the kyphoscoliotic type. Other rare forms of Ehlers-Danlos syndrome result from variants in other genes.
Some of the genes associated with the Ehlers-Danlos syndromes, including COL1A1, COL1A2, COL3A1, COL5A1, and COL5A2, provide instructions for making pieces of several different types of collagen. These pieces assemble to form mature collagen molecules that give structure and strength to connective tissues throughout the body. Other genes, including ADAMTS2, FKBP14, PLOD1, and TNXB, provide instructions for making proteins that process, fold, or interact with collagen. Variants in any of these genes disrupt the production or processing of collagen, preventing these molecules from being assembled properly. These changes weaken connective tissues in the skin, bones, and other parts of the body, resulting in the characteristic features of the Ehlers-Danlos syndromes.
Some genes associated with recently described types of Ehlers-Danlos syndrome have functions that appear to be unrelated to collagen. For many of these genes, it is not clear how variants lead to hypermobility, elastic skin, and other features of these conditions.
Inheritance
The inheritance pattern of the Ehlers-Danlos syndromes varies by type. The classical, vascular, arthrochalasia, and periodontal forms of the disorder, and likely the hypermobile type, have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means that one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the variant from one affected parent . Other cases result from new (de novo) gene variants
. Other cases result from new (de novo) gene variants and occur in people with no history of the disorder in their family.
and occur in people with no history of the disorder in their family.
The classical-like, cardiac-valvular, dermatosparaxis, kyphoscoliotic, spondylodysplastic, and musculocontractural types of Ehlers-Danlos syndrome, as well as brittle cornea syndrome, are inherited in an autosomal recessive pattern . In autosomal recessive inheritance, two copies of a gene in each cell are altered. Most often, the parents of an individual with an autosomal recessive disorder are carriers of one copy of the altered gene but do not show signs and symptoms of the disorder.
. In autosomal recessive inheritance, two copies of a gene in each cell are altered. Most often, the parents of an individual with an autosomal recessive disorder are carriers of one copy of the altered gene but do not show signs and symptoms of the disorder.
The myopathic type of Ehlers-Danlos syndrome can have either an autosomal dominant or autosomal recessive pattern of inheritance.
Other Names for This Condition
- EDS
- Ehlers Danlos disease
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Ehlers-Danlos syndrome

- Genetic Testing Registry: Ehlers-Danlos syndrome, musculocontractural type
- Genetic Testing Registry: Ehlers-Danlos syndrome, musculocontractural type 2
- Genetic Testing Registry: Ehlers-Danlos syndrome type 7A
- Genetic Testing Registry: Ehlers-Danlos syndrome type 7B
- Genetic Testing Registry: Ehlers-Danlos syndrome, cardiac valvular type
- Genetic Testing Registry: Ehlers-Danlos syndrome, dermatosparaxis type
- Genetic Testing Registry: Ehlers-Danlos syndrome, kyphoscoliotic and deafness type
- Genetic Testing Registry: Ehlers-Danlos syndrome, kyphoscoliotic type 1
- Genetic Testing Registry: Ehlers-Danlos syndrome, periodontal type 1
- Genetic Testing Registry: Ehlers-Danlos syndrome, periodontal type 2
Genetic and Rare Diseases Information Center
- Arthrochalasia Ehlers-Danlos syndrome
- Brittle cornea syndrome
- Cardiac-valvular Ehlers-Danlos syndrome
- Classical Ehlers-Danlos syndrome
- Classical-like Ehlers-Danlos syndrome type 1
- Dermatosparaxis Ehlers-Danlos syndrome
- Ehlers-Danlos syndrome
- Ehlers-Danlos syndrome, dysfibronectinemic type
- Hypermobile Ehlers-Danlos syndrome
- Kyphoscoliotic Ehlers-Danlos syndrome
- Musculocontractural Ehlers-Danlos syndrome
- Periodontal Ehlers-Danlos syndrome
- Vascular Ehlers-Danlos syndrome
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- EHLERS-DANLOS SYNDROME, CLASSIC TYPE, 1; EDSCL1
- EHLERS-DANLOS SYNDROME, CLASSIC TYPE, 2; EDSCL2
- EHLERS-DANLOS SYNDROME, HYPERMOBILITY TYPE; EDSHMB
- EHLERS-DANLOS SYNDROME, VASCULAR TYPE; EDSVASC
- EHLERS-DANLOS SYNDROME, ARTHROCHALASIA TYPE, 1; EDSARTH1
- EHLERS-DANLOS SYNDROME, SPONDYLODYSPLASTIC TYPE, 1; EDSSPD1
- EHLERS-DANLOS SYNDROME, PERIODONTAL TYPE, 1; EDSPD1
- EHLERS-DANLOS SYNDROME, AUTOSOMAL DOMINANT, TYPE UNSPECIFIED
- BRITTLE CORNEA SYNDROME 1; BCS1
- EHLERS-DANLOS SYNDROME, MUSCULOCONTRACTURAL TYPE, 1; EDSMC1
- CARDIAC VALVULAR DYSPLASIA, X-LINKED; CVDPX
- EHLERS-DANLOS SYNDROME WITH PLATELET DYSFUNCTION FROM FIBRONECTIN ABNORMALITY
- EHLERS-DANLOS SYNDROME, CARDIAC VALVULAR TYPE; EDSCV
- EHLERS-DANLOS SYNDROME, KYPHOSCOLIOTIC TYPE, 1; EDSKSCL1
- EHLERS-DANLOS SYNDROME, DERMATOSPARAXIS TYPE; EDSDERMS
- EHLERS-DANLOS SYNDROME, BEASLEY-COHEN TYPE
- EHLERS-DANLOS SYNDROME, CLASSIC-LIKE, 1; EDSCLL1
- EHLERS-DANLOS SYNDROME, SPONDYLODYSPLASTIC TYPE, 2; EDSSPD2
- EHLERS-DANLOS SYNDROME, KYPHOSCOLIOTIC TYPE, 2; EDSKSCL2
- EHLERS-DANLOS SYNDROME, MUSCULOCONTRACTURAL TYPE, 2; EDSMC2
- EHLERS-DANLOS SYNDROME, CLASSIC-LIKE, 2; EDSCLL2
- EHLERS-DANLOS SYNDROME, PERIODONTAL TYPE, 2; EDSPD2
Scientific Articles on PubMed
References
- Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998 Apr 28;77(1):31-7. doi: 10.1002/(sici)1096-8628(19980428)77:13.0.co;2-o. Citation on PubMed
- Bowen JM, Sobey GJ, Burrows NP, Colombi M, Lavallee ME, Malfait F, Francomano CA. Ehlers-Danlos syndrome, classical type. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):27-39. doi: 10.1002/ajmg.c.31548. Epub 2017 Feb 13. Citation on PubMed
- Brady AF, Demirdas S, Fournel-Gigleux S, Ghali N, Giunta C, Kapferer-Seebacher I, Kosho T, Mendoza-Londono R, Pope MF, Rohrbach M, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Zschocke J, Malfait F. The Ehlers-Danlos syndromes, rare types. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):70-115. doi: 10.1002/ajmg.c.31550. Citation on PubMed
- Byers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X, Johnson D, Pepin M, Robert L, Sanders L, Wheeldon N. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):40-47. doi: 10.1002/ajmg.c.31553. Citation on PubMed
- Byers PH. Vascular Ehlers-Danlos Syndrome. 1999 Sep 2 [updated 2019 Feb 21]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1494/ Citation on PubMed
- Chopra P, Tinkle B, Hamonet C, Brock I, Gompel A, Bulbena A, Francomano C. Pain management in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):212-219. doi: 10.1002/ajmg.c.31554. Epub 2017 Feb 10. Citation on PubMed
- Engelbert RH, Juul-Kristensen B, Pacey V, de Wandele I, Smeenk S, Woinarosky N, Sabo S, Scheper MC, Russek L, Simmonds JV. The evidence-based rationale for physical therapy treatment of children, adolescents, and adults diagnosed with joint hypermobility syndrome/hypermobile Ehlers Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):158-167. doi: 10.1002/ajmg.c.31545. Citation on PubMed
- Ericson WB Jr, Wolman R. Orthopaedic management of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):188-194. doi: 10.1002/ajmg.c.31551. Epub 2017 Feb 13. Citation on PubMed
- Hakim A. Hypermobile Ehlers-Danlos Syndrome. 2004 Oct 22 [updated 2024 Feb 22]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1279/ Citation on PubMed
- Jesudas R, Chaudhury A, Laukaitis CM. An update on the new classification of Ehlers-Danlos syndrome and review of the causes of bleeding in this population. Haemophilia. 2019 Jul;25(4):558-566. doi: 10.1111/hae.13800. Epub 2019 Jun 10. Citation on PubMed
- Malfait F, De Paepe A. The Ehlers-Danlos syndrome. Adv Exp Med Biol. 2014;802:129-43. doi: 10.1007/978-94-007-7893-1_9. Citation on PubMed
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):8-26. doi: 10.1002/ajmg.c.31552. Citation on PubMed
- Malfait F, Symoens S, Syx D. Classic Ehlers-Danlos Syndrome. 2007 May 29 [updated 2024 Feb 1]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1244/ Citation on PubMed
- Tinkle B, Castori M, Berglund B, Cohen H, Grahame R, Kazkaz H, Levy H. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):48-69. doi: 10.1002/ajmg.c.31538. Epub 2017 Feb 1. Citation on PubMed
- van Dijk FS, Ghali N, Demirdas S, Baker D. TNXB-Related Classical-Like Ehlers-Danlos Syndrome. 2022 Sep 15. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK584019/ Citation on PubMed
- Yeowell HN, Steinmann B. PLOD1-Related Kyphoscoliotic Ehlers-Danlos Syndrome. 2000 Feb 2 [updated 2018 Oct 18]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1462/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.