

Description
Dyskeratosis congenita is a disorder that can affect many parts of the body. There are three features that are characteristic of this disorder: fingernails and toenails that grow poorly or are abnormally shaped (nail dystrophy); changes in skin coloring (pigmentation), especially on the neck and chest, in a pattern often described as "lacy"; and white patches inside the mouth (oral leukoplakia).

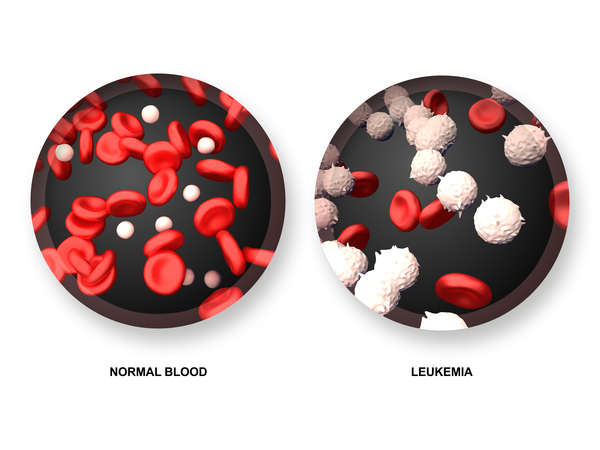
People with dyskeratosis congenita have an increased risk of developing several life-threatening conditions. They are especially vulnerable to disorders that impair bone marrow function. These disorders disrupt the ability of the bone marrow to produce new blood cells. Affected individuals may develop aplastic anemia, also known as bone marrow failure, which occurs when the bone marrow does not produce enough new blood cells. They are also at higher than average risk for myelodysplastic syndrome, a condition in which immature blood cells fail to develop normally; this condition may progress to a form of blood cancer called leukemia. People with dyskeratosis congenita are also at increased risk of developing leukemia even if they never develop myelodysplastic syndrome. In addition, they have a higher than average risk of developing other cancers, especially cancers of the head, neck, anus, or genitals.
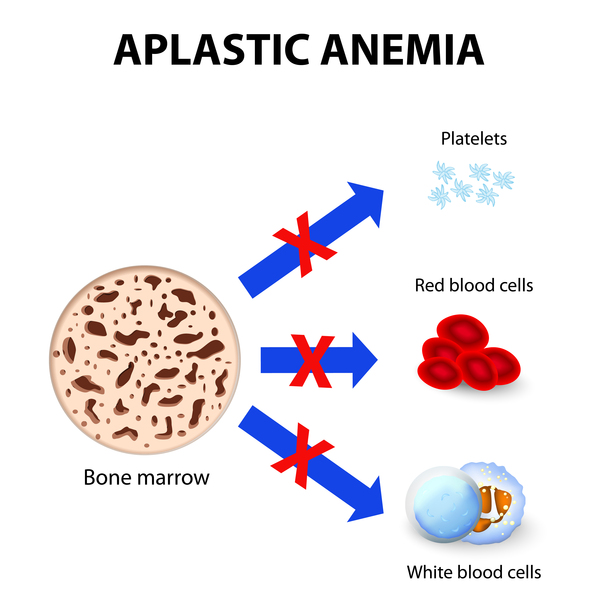
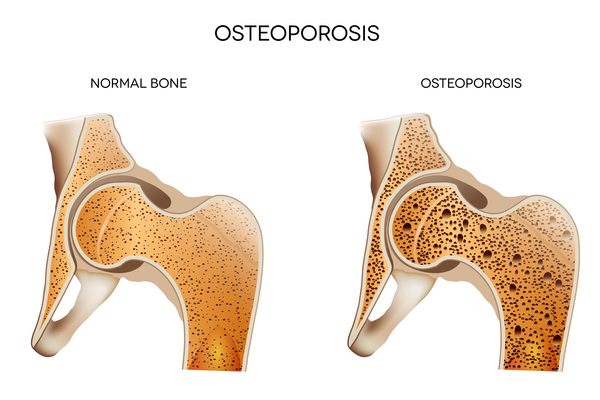
People with dyskeratosis congenita may also develop pulmonary fibrosis, a condition that causes scar tissue (fibrosis) to build up in the lungs, decreasing the transport of oxygen into the bloodstream. Additional signs and symptoms that occur in some people with dyskeratosis congenita include eye abnormalities such as narrow tear ducts that may become blocked, preventing drainage of tears and leading to eyelid irritation; dental problems; hair loss or prematurely grey hair; low bone mineral density (osteoporosis); degeneration (avascular necrosis) of the hip and shoulder joints; or liver disease. Some affected males may have narrowing (stenosis) of the urethra, which is the tube that carries urine out of the body from the bladder. Urethral stenosis may lead to difficult or painful urination and urinary tract infections.
The severity of dyskeratosis congenita varies widely among affected individuals. The least severely affected individuals have only a few mild physical features of the disorder and normal bone marrow function. More severely affected individuals have many of the characteristic physical features and experience bone marrow failure, cancer, or pulmonary fibrosis by early adulthood.
While most people with dyskeratosis congenita have normal intelligence and development of motor skills such as standing and walking, developmental delay may occur in some severely affected individuals. In one severe form of the disorder called Hoyeraal Hreidaarsson syndrome, affected individuals have an unusually small and underdeveloped cerebellum, which is the part of the brain that coordinates movement. Another severe variant called Revesz syndrome involves abnormalities in the light-sensitive tissue at the back of the eye (retina) in addition to the other symptoms of dyskeratosis congenita.


Frequency
The exact prevalence of dyskeratosis congenita is unknown. It is estimated to occur in approximately 1 in 1 million people.
Causes
In about half of people with dyskeratosis congenita, the disorder is caused by mutations in the TERT, TERC, DKC1, or TINF2 gene. These genes provide instructions for making proteins that help maintain structures known as telomeres, which are found at the ends of chromosomes. In a small number of individuals with dyskeratosis congenita, mutations in other genes involved with telomere maintenance have been identified. Other affected individuals have no mutations in any of the genes currently associated with dyskeratosis congenita. In these cases, the cause of the disorder is unknown, but other unidentified genes related to telomere maintenance are likely involved.
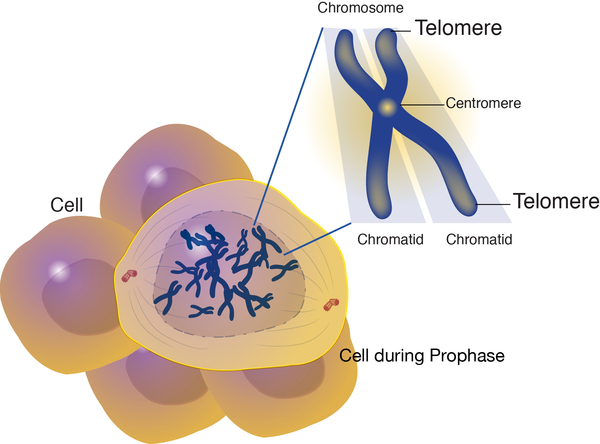
Telomeres help protect chromosomes from abnormally sticking together or breaking down (degrading). In most cells, telomeres become progressively shorter as the cell divides. After a certain number of cell divisions, the telomeres become so short that they trigger the cell to stop dividing or to self-destruct (undergo apoptosis).
Telomeres are maintained by two important protein complexes called telomerase and shelterin. Telomerase helps maintain normal telomere length by adding small repeated segments of DNA to the ends of chromosomes each time the cell divides. The main components of telomerase, called hTR and hTERT, are produced from the TERC and TERT genes, respectively. The hTR component is an RNA molecule, a chemical cousin of DNA. It provides a template for creating the repeated sequence of DNA that telomerase adds to the ends of chromosomes. The function of the hTERT component is to add the new DNA segment to chromosome ends. The DKC1 gene provides instructions for making another protein that is important in telomerase function. This protein, called dyskerin, attaches (binds) to hTR and helps stabilize the telomerase complex.
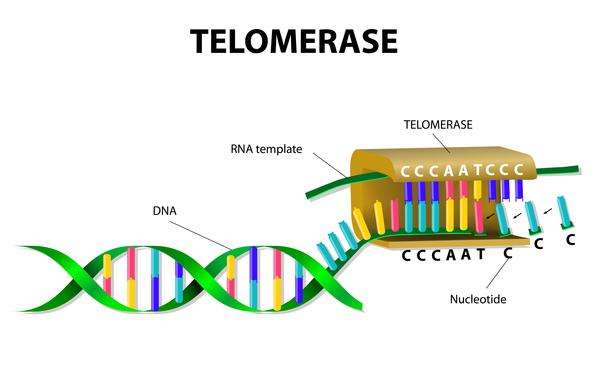
The shelterin complex helps protect telomeres from the cell's DNA repair process. Without the protection of shelterin, the repair mechanism would sense the chromosome ends as abnormal breaks in the DNA sequence and either attempt to join the ends together or initiate apoptosis. The TINF2 gene provides instructions for making a protein that is part of the shelterin complex.
TERT, TERC, DKC1, or TINF2 gene mutations result in dysfunction of the telomerase or shelterin complexes, leading to impaired maintenance of telomeres and reduced telomere length. Cells that divide rapidly are especially vulnerable to the effects of shortened telomeres. As a result, people with dyskeratosis congenita may experience a variety of problems affecting quickly dividing cells in the body such as cells of the nail beds, hair follicles, skin, lining of the mouth (oral mucosa), and bone marrow.
Breakage and instability of chromosomes resulting from inadequate telomere maintenance may lead to genetic changes that allow cells to divide in an uncontrolled way, resulting in the development of cancer in people with dyskeratosis congenita.
Inheritance
Dyskeratosis congenita can have different inheritance patterns.
When dyskeratosis congenita is caused by DKC1 gene mutations, it is inherited in an X-linked recessive pattern. The DKC1 gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

When dyskeratosis congenita is caused by mutations in other genes, it can be inherited in an autosomal dominant or autosomal recessive pattern. Autosomal dominant means one copy of the altered gene in each cell is sufficient to cause the disorder. Autosomal recessive means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.


Other Names for This Condition
- Zinsser-Cole-Engman syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- DYSKERATOSIS CONGENITA, AUTOSOMAL DOMINANT 1; DKCA1
- DYSKERATOSIS CONGENITA, AUTOSOMAL RECESSIVE 1; DKCB1
- DYSKERATOSIS CONGENITA, X-LINKED; DKCX
- REVESZ SYNDROME
- DYSKERATOSIS CONGENITA, AUTOSOMAL RECESSIVE 2; DKCB2
- DYSKERATOSIS CONGENITA, AUTOSOMAL RECESSIVE 3; DKCB3
- DYSKERATOSIS CONGENITA, AUTOSOMAL DOMINANT 2; DKCA2
- DYSKERATOSIS CONGENITA, AUTOSOMAL DOMINANT 3; DKCA3
- DYSKERATOSIS CONGENITA, AUTOSOMAL RECESSIVE 5; DKCB5
Scientific Articles on PubMed
References
- Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009 Jun 25;113(26):6549-57. doi: 10.1182/blood-2008-12-192880. Epub 2009 Mar 12. Citation on PubMed or Free article on PubMed Central
- Ballew BJ, Savage SA. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2013 Jun;6(3):327-37. doi: 10.1586/ehm.13.23. Citation on PubMed
- Baran I, Nalcaci R, Kocak M. Dyskeratosis congenita: clinical report and review of the literature. Int J Dent Hyg. 2010 Feb;8(1):68-74. doi: 10.1111/j.1601-5037.2009.00364.x. Citation on PubMed
- Calado RT. Telomeres and marrow failure. Hematology Am Soc Hematol Educ Program. 2009:338-43. doi: 10.1182/asheducation-2009.1.338. Citation on PubMed
- Keller RB, Gagne KE, Usmani GN, Asdourian GK, Williams DA, Hofmann I, Agarwal S. CTC1 Mutations in a patient with dyskeratosis congenita. Pediatr Blood Cancer. 2012 Aug;59(2):311-4. doi: 10.1002/pbc.24193. Epub 2012 Apr 24. Citation on PubMed or Free article on PubMed Central
- Kirwan M, Dokal I. Dyskeratosis congenita, stem cells and telomeres. Biochim Biophys Acta. 2009 Apr;1792(4):371-9. doi: 10.1016/j.bbadis.2009.01.010. Epub 2009 Feb 7. Citation on PubMed or Free article on PubMed Central
- Le Guen T, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, Carpentier W, Nitschke P, Picard C, Couillault G, Soulier J, Fischer A, Callebaut I, Jabado N, Londono-Vallejo A, de Villartay JP, Revy P. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013 Aug 15;22(16):3239-49. doi: 10.1093/hmg/ddt178. Epub 2013 Apr 15. Citation on PubMed
- Nishio N, Kojima S. Recent progress in dyskeratosis congenita. Int J Hematol. 2010 Oct;92(3):419-24. doi: 10.1007/s12185-010-0695-5. Epub 2010 Oct 1. Citation on PubMed
- Savage SA, Niewisch MR. Dyskeratosis Congenita and Related Telomere Biology Disorders. 2009 Nov 12 [updated 2023 Jan 19]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK22301/ Citation on PubMed
- Walne AJ, Bhagat T, Kirwan M, Gitiaux C, Desguerre I, Leonard N, Nogales E, Vulliamy T, Dokal IS. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica. 2013 Mar;98(3):334-8. doi: 10.3324/haematol.2012.071068. Epub 2012 Aug 16. Citation on PubMed or Free article on PubMed Central
- Walne AJ, Dokal I. Advances in the understanding of dyskeratosis congenita. Br J Haematol. 2009 Apr;145(2):164-72. doi: 10.1111/j.1365-2141.2009.07598.x. Epub 2009 Feb 4. Citation on PubMed or Free article on PubMed Central
- Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T, Chen R, Alter BP, Artandi SE. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011 Jan 1;25(1):11-6. doi: 10.1101/gad.2006411. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.