

Description
Dihydropyrimidinase deficiency is a disorder that can cause neurological and gastrointestinal problems in some affected individuals. Other people with dihydropyrimidinase deficiency have no signs or symptoms related to the disorder, and in these individuals the condition can be diagnosed only by laboratory testing.
The neurological abnormalities that occur most often in people with dihydropyrimidinase deficiency are intellectual disability, seizures, and weak muscle tone (hypotonia). An abnormally small head size (microcephaly) and autistic behaviors that affect communication and social interaction also occur in some individuals with this condition.

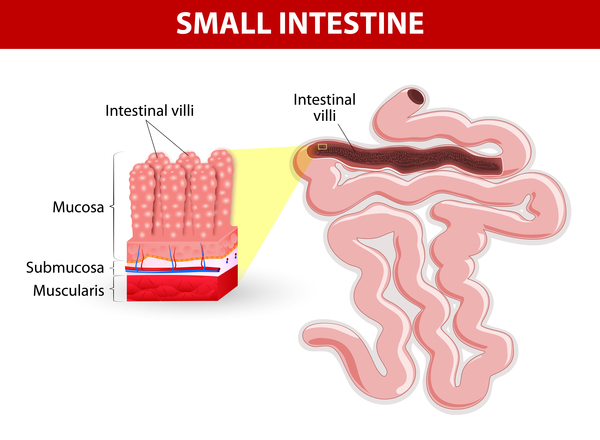
Gastrointestinal problems that occur in dihydropyrimidinase deficiency include backflow of acidic stomach contents into the esophagus (gastroesophageal reflux) and recurrent episodes of vomiting (cyclic vomiting). Affected individuals can also have deterioration (atrophy) of the small, finger-like projections (villi) that line the small intestine and provide a large surface area with which to absorb nutrients. This condition, called villous atrophy, can lead to difficulty absorbing nutrients from foods (malabsorption), resulting in a failure to grow and gain weight at the expected rate (failure to thrive).
People with dihydropyrimidinase deficiency, including those who otherwise exhibit no symptoms, may be vulnerable to severe, potentially life-threatening toxic reactions to certain drugs called fluoropyrimidines that are used to treat cancer. Common examples of these drugs are 5-fluorouracil and capecitabine. These drugs may not be broken down efficiently and can build up to toxic levels in the body (fluoropyrimidine toxicity), leading to drug reactions including gastrointestinal problems, blood abnormalities, and other signs and symptoms.
Frequency
Dihydropyrimidinase deficiency is thought to be a rare disorder. Only a few dozen affected individuals have been described in the medical literature.
Causes
Dihydropyrimidinase deficiency is caused by mutations in the DPYS gene, which provides instructions for making an enzyme called dihydropyrimidinase. This enzyme is involved in the breakdown of molecules called pyrimidines, which are building blocks of DNA and its chemical cousin RNA. The dihydropyrimidinase enzyme is involved in the second step of the three-step process that breaks down pyrimidines. This step opens the ring-like structures of molecules called 5,6-dihydrothymine and 5,6-dihydrouracil so that these molecules can be further broken down.
The DPYS gene mutations that cause dihydropyrimidinase deficiency greatly reduce or eliminate dihydropyrimidinase enzyme function. As a result, the enzyme is unable to begin the breakdown of 5,6-dihydrothymine and 5,6-dihydrouracil. Excessive amounts of these molecules accumulate in the blood and in the fluid that surrounds and protects the brain and spinal cord (the cerebrospinal fluid or CSF) and are released in the urine.
The relationship between the inability to break down 5,6-dihydrothymine and 5,6-dihydrouracil and the specific features of dihydropyrimidinase deficiency is unclear. Failure to complete this step in the breakdown of pyrimidines also impedes the final step of the process, which produces molecules called beta-aminoisobutyric acid and beta-alanine. Both of these molecules are thought to protect the nervous system and help it function properly. Reduced production of beta-aminoisobutyric acid and beta-alanine may impair the function of these molecules in the nervous system, leading to neurological problems in some people with dihydropyrimidinase deficiency. Because fluoropyrimidine drugs are broken down by the same three-step process as pyrimidines, deficiency of the dihydropyrimidinase enzyme could lead to the drug buildup that causes fluoropyrimidine toxicity.
It is unknown why some people with dihydropyrimidinase deficiency do not develop health problems related to the condition; other genetic and environmental factors likely help determine the effects of this disorder.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Dihydropyrimidinuria
- Dihydrouracil amidohydrolase deficiency
- DPH deficiency
- DPYS deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Hamajima N, Kouwaki M, Vreken P, Matsuda K, Sumi S, Imaeda M, Ohba S, Kidouchi K, Nonaka M, Sasaki M, Tamaki N, Endo Y, De Abreu R, Rotteveel J, van Kuilenburg A, van Gennip A, Togari H, Wada Y. Dihydropyrimidinase deficiency: structural organization, chromosomal localization, and mutation analysis of the human dihydropyrimidinase gene. Am J Hum Genet. 1998 Sep;63(3):717-26. doi: 10.1086/302022. Citation on PubMed or Free article on PubMed Central
- Schnackerz KD, Dobritzsch D. Amidohydrolases of the reductive pyrimidine catabolic pathway purification, characterization, structure, reaction mechanisms and enzyme deficiency. Biochim Biophys Acta. 2008 Mar;1784(3):431-44. doi: 10.1016/j.bbapap.2008.01.005. Epub 2008 Jan 18. Citation on PubMed
- Sumi S, Imaeda M, Kidouchi K, Ohba S, Hamajima N, Kodama K, Togari H, Wada Y. Population and family studies of dihydropyrimidinuria: prevalence, inheritance mode, and risk of fluorouracil toxicity. Am J Med Genet. 1998 Jul 24;78(4):336-40. doi: 10.1002/(sici)1096-8628(19980724)78:43.0.co;2-j. Citation on PubMed
- van Kuilenburg AB, Dobritzsch D, Meijer J, Meinsma R, Benoist JF, Assmann B, Schubert S, Hoffmann GF, Duran M, de Vries MC, Kurlemann G, Eyskens FJ, Greed L, Sass JO, Schwab KO, Sewell AC, Walter J, Hahn A, Zoetekouw L, Ribes A, Lind S, Hennekam RC. Dihydropyrimidinase deficiency: Phenotype, genotype and structural consequences in 17 patients. Biochim Biophys Acta. 2010 Jul-Aug;1802(7-8):639-48. doi: 10.1016/j.bbadis.2010.03.013. Epub 2010 Apr 1. Citation on PubMed
- van Kuilenburg AB, Meijer J, Dobritzsch D, Meinsma R, Duran M, Lohkamp B, Zoetekouw L, Abeling NG, van Tinteren HL, Bosch AM. Clinical, biochemical and genetic findings in two siblings with a dihydropyrimidinase deficiency. Mol Genet Metab. 2007 Jun;91(2):157-64. doi: 10.1016/j.ymgme.2007.02.008. Epub 2007 Mar 26. Citation on PubMed
- van Kuilenburg AB, Meinsma R, Zonnenberg BA, Zoetekouw L, Baas F, Matsuda K, Tamaki N, van Gennip AH. Dihydropyrimidinase deficiency and severe 5-fluorouracil toxicity. Clin Cancer Res. 2003 Oct 1;9(12):4363-7. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.