

Description
Dentatorubral-pallidoluysian atrophy (DRPLA) is a progressive brain disorder that causes involuntary movements, mental and emotional problems, and a decline in thinking ability. The average age of onset for DRPLA is around 30 years, but this condition can appear any time between infancy and mid-adulthood.
The signs and symptoms of DRPLA differ somewhat between affected children and adults. When DRPLA appears before age 20, it most often involves episodes of involuntary muscle jerking or twitching (myoclonus), seizures, behavioral changes, intellectual disabilities, and problems with balance and coordination (ataxia). When DRPLA begins after age 20, the most frequent signs and symptoms are ataxia, uncontrollable movements of the limbs (choreoathetosis), psychiatric symptoms such as delusions, and deterioration of intellectual function (dementia).
Frequency
DRPLA is most common in the Japanese population, where it is estimated to affect 2 to 7 per million people. However, this condition has also been seen in families around world.
Although DRPLA is rare in the United States, it has been studied in a large African American family from the Haw River area of North Carolina. When the family was first identified, researchers named the disorder Haw River syndrome. Later, researchers determined that Haw River syndrome and DRPLA are the same condition.
Causes
DRPLA is caused by a variant (also called mutation) in the ATN1 gene. This gene provides instructions for making a protein called atrophin 1. Although the exact function of atrophin 1 is unknown, it appears to play an important role in nerve cells (neurons) in many areas of the brain.
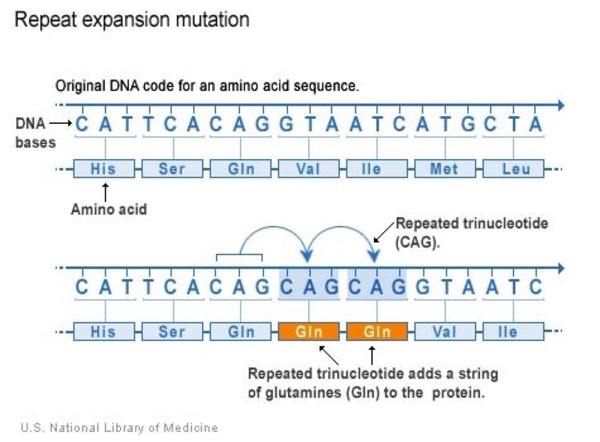
The ATN1 gene variant that underlies DRPLA involves a DNA segment known as a CAG trinucleotide repeat. This segment is made up of a series of three DNA building blocks (cytosine, adenine, and guanine) that appear multiple times in a row. Normally, this segment is repeated 6 to 35 times within the ATN1 gene. In people with DRPLA, the CAG segment is repeated at least 48 times, and the repeat region may be two or three times its usual length. The abnormally long CAG trinucleotide repeat changes the structure of the atrophin 1 protein. This altered protein accumulates in neurons and interferes with normal cell functions. The dysfunction and eventual death of these neurons lead to uncontrolled movements, intellectual decline, and the other characteristic features of DRPLA.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition.

As the altered ATN1 gene is passed from one generation to the next, the CAG trinucleotide repeat may increase in size. A minor increase in length may lead to mild symptoms or even no symptoms at all. Larger increases are usually associated with an earlier onset of the disorder and more severe signs and symptoms. This phenomenon is called anticipation. Anticipation tends to be more prominent when the altered ATN1 gene is inherited from a person's father (paternal inheritance) than when it is inherited from a person's mother (maternal inheritance).
Other Names for This Condition
- DRPLA
- Haw River syndrome
- Myoclonic epilepsy with choreoathetosis
- Naito-Oyanagi disease
- NOD
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Burke JR, Wingfield MS, Lewis KE, Roses AD, Lee JE, Hulette C, Pericak-Vance MA, Vance JM. The Haw River syndrome: dentatorubropallidoluysian atrophy (DRPLA) in an African-American family. Nat Genet. 1994 Aug;7(4):521-4. doi: 10.1038/ng0894-521. Citation on PubMed
- Chaudhry A, Anthanasiou-Fragkouli A, Houlden H. DRPLA: understanding the natural history and developing biomarkers to accelerate therapeutic trials in a globally rare repeat expansion disorder. J Neurol. 2021 Aug;268(8):3031-3041. doi: 10.1007/s00415-020-10218-6. Epub 2020 Oct 26. Erratum In: J Neurol. 2021 Jun 21;: Citation on PubMed
- Ikeuchi T, Koide R, Onodera O, Tanaka H, Oyake M, Takano H, Tsuji S. Dentatorubral-pallidoluysian atrophy (DRPLA). Molecular basis for wide clinical features of DRPLA. Clin Neurosci. 1995;3(1):23-7. Citation on PubMed
- Ikeuchi T, Onodera O, Oyake M, Koide R, Tanaka H, Tsuji S. Dentatorubral-pallidoluysian atrophy (DRPLA): close correlation of CAG repeat expansions with the wide spectrum of clinical presentations and prominent anticipation. Semin Cell Biol. 1995 Feb;6(1):37-44. doi: 10.1016/1043-4682(95)90013-6. Citation on PubMed
- Kanazawa I. Dentatorubral-pallidoluysian atrophy or Naito-Oyanagi disease. Neurogenetics. 1998 Dec;2(1):1-17. doi: 10.1007/s100480050046. Citation on PubMed
- Maruyama S, Saito Y, Nakagawa E, Saito T, Komaki H, Sugai K, Sasaki M, Kumada S, Saito Y, Tanaka H, Minami N, Goto Y. Importance of CAG repeat length in childhood-onset dentatorubral-pallidoluysian atrophy. J Neurol. 2012 Nov;259(11):2329-34. doi: 10.1007/s00415-012-6493-7. Epub 2012 Apr 18. Citation on PubMed
- Nagafuchi S, Yanagisawa H, Ohsaki E, Shirayama T, Tadokoro K, Inoue T, Yamada M. Structure and expression of the gene responsible for the triplet repeat disorder, dentatorubral and pallidoluysian atrophy (DRPLA). Nat Genet. 1994 Oct;8(2):177-82. doi: 10.1038/ng1094-177. Citation on PubMed
- Nowak B, Kozlowska E, Pawlik W, Fiszer A. Atrophin-1 Function and Dysfunction in Dentatorubral-Pallidoluysian Atrophy. Mov Disord. 2023 Apr;38(4):526-536. doi: 10.1002/mds.29355. Epub 2023 Feb 21. Citation on PubMed
- Prades S PhD, Melo de Gusmao C MD, Grimaldi S MD, Shiloh-Malawsky Y MD, Felton T MS, CGC, Houlden H MD, PhD. DRPLA. 1999 Aug 6 [updated 2023 Sep 21]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1491/ Citation on PubMed
- Wardle M, Morris HR, Robertson NP. Clinical and genetic characteristics of non-Asian dentatorubral-pallidoluysian atrophy: A systematic review. Mov Disord. 2009 Aug 15;24(11):1636-40. doi: 10.1002/mds.22642. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.