

Description
D-bifunctional protein deficiency is a disorder that causes deterioration of nervous system functions (neurodegeneration) beginning in infancy. Newborns with D-bifunctional protein deficiency have weak muscle tone (hypotonia) and seizures. Most babies with this condition never acquire any developmental skills. Some may reach very early developmental milestones such as the ability to follow movement with their eyes or control their head movement, but they experience a gradual loss of these skills (developmental regression) within a few months. As the condition gets worse, affected children develop exaggerated reflexes (hyperreflexia), increased muscle tone (hypertonia), more severe and recurrent seizures (epilepsy), and loss of vision and hearing. Most children with D-bifunctional protein deficiency do not survive past the age of 2. A small number of individuals with this disorder are somewhat less severely affected. They may acquire additional basic skills, such as voluntary hand movements or unsupported sitting, before experiencing developmental regression, and they may survive longer into childhood than more severely affected individuals.
Individuals with D-bifunctional protein deficiency may have unusual facial features, including a high forehead, widely spaced eyes (hypertelorism ), a lengthened area between the nose and mouth (philtrum), and a high arch of the hard palate
), a lengthened area between the nose and mouth (philtrum), and a high arch of the hard palate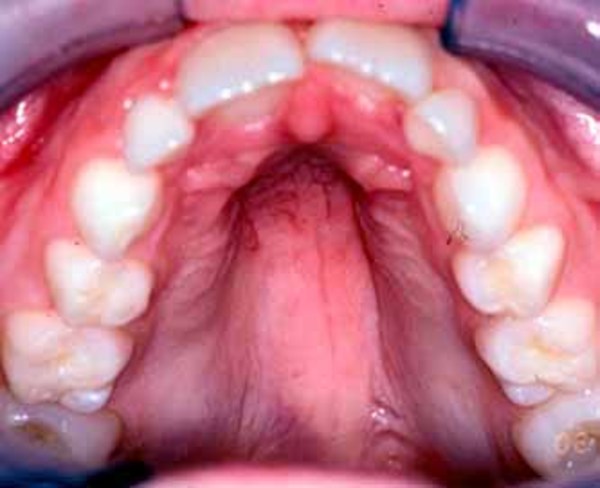 at the roof of the mouth. Affected infants may also have an unusually large space between the bones of the skull (fontanelle). An enlarged liver
at the roof of the mouth. Affected infants may also have an unusually large space between the bones of the skull (fontanelle). An enlarged liver (hepatomegaly) occurs in about half of affected individuals. Because these features are similar to those of another disorder called Zellweger syndrome (part of a group of disorders called the Zellweger spectrum), D-bifunctional protein deficiency is sometimes called pseudo-Zellweger syndrome.
(hepatomegaly) occurs in about half of affected individuals. Because these features are similar to those of another disorder called Zellweger syndrome (part of a group of disorders called the Zellweger spectrum), D-bifunctional protein deficiency is sometimes called pseudo-Zellweger syndrome.
Frequency
D-bifunctional protein deficiency is estimated to affect 1 in 100,000 newborns.
Causes
D-bifunctional protein deficiency is caused by variants (also known as mutations) in the HSD17B4 gene. The protein produced from this gene (D-bifunctional protein) is an enzyme, which means that it helps specific biochemical reactions to take place. The D-bifunctional protein is found in sac-like cell structures (organelles) called peroxisomes , which contain a variety of enzymes that break down many different substances. The D-bifunctional protein is involved in the breakdown of certain molecules called fatty acids. The protein has two separate regions (domains) with enzyme activity, called the hydratase and dehydrogenase domains. These domains help carry out the second and third steps, respectively, of a process called the peroxisomal fatty acid beta-oxidation pathway. This process shortens the fatty acid molecules by two carbon atoms at a time until the fatty acids are converted to a molecule called acetyl-CoA, which is transported out of the peroxisomes for reuse by the cell.
, which contain a variety of enzymes that break down many different substances. The D-bifunctional protein is involved in the breakdown of certain molecules called fatty acids. The protein has two separate regions (domains) with enzyme activity, called the hydratase and dehydrogenase domains. These domains help carry out the second and third steps, respectively, of a process called the peroxisomal fatty acid beta-oxidation pathway. This process shortens the fatty acid molecules by two carbon atoms at a time until the fatty acids are converted to a molecule called acetyl-CoA, which is transported out of the peroxisomes for reuse by the cell.
HSD17B4 gene variants that cause D-bifunctional protein deficiency can affect one or both of the protein's functions; however, this distinction does not seem to affect the severity or features of the disorder. Impairment of one or both of the protein's enzymatic activities prevents the D-bifunctional protein from breaking down fatty acids efficiently. As a result, these fatty acids accumulate in the body. It is unclear how fatty acid accumulation leads to the specific neurological and non-neurological features of D-bifunctional protein deficiency. However, the accumulation may result in abnormal development of the brain and the breakdown of myelin, which is the covering that protects nerves and promotes the efficient transmission of nerve impulses. Destruction of myelin leads to a loss of myelin-containing tissue (white matter) in the brain and spinal cord; loss of white matter is described as leukodystrophy. Abnormal brain development and leukodystrophy likely underlie the neurological abnormalities that occur in D-bifunctional protein deficiency.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- 17-beta-hydroxysteroid dehydrogenase IV deficiency
- Bifunctional peroxisomal enzyme deficiency
- DBP deficiency
- PBFE deficiency
- Peroxisomal bifunctional enzyme deficiency
- Pseudo-Zellweger syndrome
- Zellweger-like syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ferdinandusse S, Denis S, Mooyer PA, Dekker C, Duran M, Soorani-Lunsing RJ, Boltshauser E, Macaya A, Gartner J, Majoie CB, Barth PG, Wanders RJ, Poll-The BT. Clinical and biochemical spectrum of D-bifunctional protein deficiency. Ann Neurol. 2006 Jan;59(1):92-104. doi: 10.1002/ana.20702. Citation on PubMed
- Ferdinandusse S, Ylianttila MS, Gloerich J, Koski MK, Oostheim W, Waterham HR, Hiltunen JK, Wanders RJ, Glumoff T. Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis. Am J Hum Genet. 2006 Jan;78(1):112-24. doi: 10.1086/498880. Epub 2005 Nov 15. Citation on PubMed or Free article on PubMed Central
- McMillan HJ, Worthylake T, Schwartzentruber J, Gottlieb CC, Lawrence SE, Mackenzie A, Beaulieu CL, Mooyer PA; FORGE Canada Consortium; Wanders RJ, Majewski J, Bulman DE, Geraghty MT, Ferdinandusse S, Boycott KM. Specific combination of compound heterozygous mutations in 17beta-hydroxysteroid dehydrogenase type 4 (HSD17B4) defines a new subtype of D-bifunctional protein deficiency. Orphanet J Rare Dis. 2012 Nov 22;7:90. doi: 10.1186/1750-1172-7-90. Citation on PubMed or Free article on PubMed Central
- Moller G, van Grunsven EG, Wanders RJ, Adamski J. Molecular basis of D-bifunctional protein deficiency. Mol Cell Endocrinol. 2001 Jan 22;171(1-2):61-70. doi: 10.1016/s0303-7207(00)00388-9. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.