

Description
Cystinuria is a condition characterized by the buildup of the amino acid cystine, a building block of most proteins, in the kidneys and bladder. As the kidneys filter blood to create urine, cystine is normally absorbed back into the bloodstream. People with cystinuria cannot properly reabsorb cystine into their bloodstream, so the amino acid accumulates in their urine.

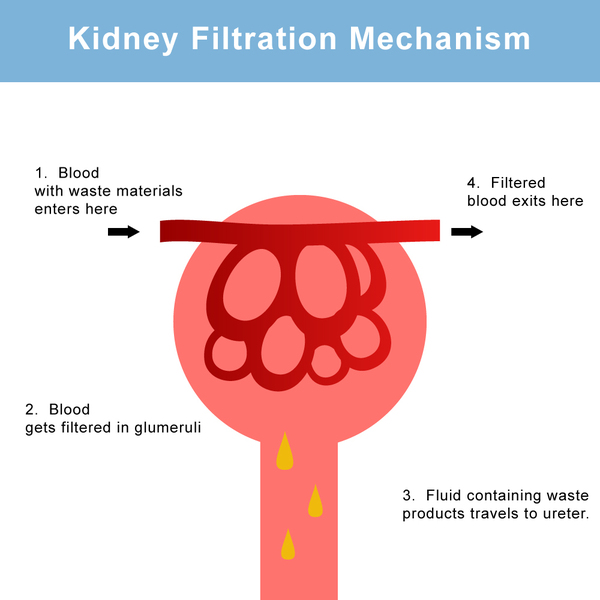
As urine becomes more concentrated in the kidneys, the excess cystine forms crystals. Larger crystals become stones that may lodge in the kidneys or in the bladder. Sometimes cystine crystals combine with calcium molecules in the kidneys to form large stones. These crystals and stones can create blockages in the urinary tract and reduce the ability of the kidneys to eliminate waste through urine. The stones also provide sites where bacteria may cause infections.
Frequency
Cystinuria affects approximately 1 in 10,000 people.
Causes
Mutations in the SLC3A1 or SLC7A9 gene cause cystinuria. The SLC3A1 and SLC7A9 genes provide instructions for making the two parts (subunits) of a protein complex that is primarily found in the kidneys. Normally this protein complex controls the reabsorption of certain amino acids, including cystine, into the blood from the filtered fluid that will become urine. Mutations in either the SLC3A1 gene or SLC7A9 gene disrupt the ability of the protein complex to reabsorb amino acids, which causes the amino acids to become concentrated in the urine. As the levels of cystine in the urine increase, the crystals typical of cystinuria form. The other amino acids that are reabsorbed by the protein complex do not create crystals when they accumulate in the urine.

Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- CSNU
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Claes DJ, Jackson E. Cystinuria: mechanisms and management. Pediatr Nephrol. 2012 Nov;27(11):2031-2038. doi: 10.1007/s00467-011-2092-6. Epub 2012 Jan 27. Citation on PubMed
- Knoll T, Zollner A, Wendt-Nordahl G, Michel MS, Alken P. Cystinuria in childhood and adolescence: recommendations for diagnosis, treatment, and follow-up. Pediatr Nephrol. 2005 Jan;20(1):19-24. doi: 10.1007/s00467-004-1663-1. Epub 2004 Nov 25. Citation on PubMed
- Langman CB. The molecular basis of kidney stones. Curr Opin Pediatr. 2004 Apr;16(2):188-93. doi: 10.1097/00008480-200404000-00013. Citation on PubMed
- Mattoo A, Goldfarb DS. Cystinuria. Semin Nephrol. 2008 Mar;28(2):181-91. doi: 10.1016/j.semnephrol.2008.01.011. Citation on PubMed
- Saravakos P, Kokkinou V, Giannatos E. Cystinuria: current diagnosis and management. Urology. 2014 Apr;83(4):693-9. doi: 10.1016/j.urology.2013.10.013. Epub 2013 Nov 16. Citation on PubMed
- Townsend DM, Tew KD, Tapiero H. Sulfur containing amino acids and human disease. Biomed Pharmacother. 2004 Jan;58(1):47-55. doi: 10.1016/j.biopha.2003.11.005. Citation on PubMed
- Trinchieri A, Dormia G, Montanari E, Zanetti G. Cystinuria: definition, epidemiology and clinical aspects. Arch Ital Urol Androl. 2004 Sep;76(3):129-34. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.