

Description
Cystic fibrosis is an inherited disease characterized by the buildup of thick, sticky mucus that can damage many of the body's organs. The disorder's most common signs and symptoms include progressive damage to the respiratory system and chronic digestive system problems. The features of the disorder and their severity varies among affected individuals.
Mucus is a slippery substance that lubricates and protects the linings of the airways, digestive system, reproductive system, and other organs and tissues. In people with cystic fibrosis, the body produces mucus that is abnormally thick and sticky. This abnormal mucus can clog the airways , leading to severe problems with breathing and bacterial infections in the lungs. These infections cause chronic coughing, wheezing, and inflammation. Over time, mucus buildup and infections result in permanent lung damage, including the formation of scar tissue (fibrosis) and cysts in the lungs.
, leading to severe problems with breathing and bacterial infections in the lungs. These infections cause chronic coughing, wheezing, and inflammation. Over time, mucus buildup and infections result in permanent lung damage, including the formation of scar tissue (fibrosis) and cysts in the lungs.
Most people with cystic fibrosis also have digestive problems. Some affected babies have meconium ileus, a blockage of the intestine that occurs shortly after birth. Other digestive problems result from a buildup of thick, sticky mucus in the pancreas. The pancreas is an organ that produces insulin (a hormone that helps control blood glucose levels). It also makes enzymes that help digest food. In people with cystic fibrosis, mucus often damages the pancreas, impairing its ability to produce insulin and digestive enzymes. Problems with digestion can lead to diarrhea, malnutrition, poor growth, and weight loss. In adolescence or adulthood, a shortage of insulin can cause a form of diabetes known as cystic fibrosis-related diabetes mellitus (CFRDM).
Cystic fibrosis used to be considered a fatal disease of childhood. With improved treatments and better ways to manage the disease, many people with cystic fibrosis now live well into adulthood. Adults with cystic fibrosis experience health problems affecting the respiratory, digestive, and reproductive systems. Most men with cystic fibrosis have congenital bilateral absence of the vas deferens (CBAVD), a condition in which the tubes that carry sperm (the vas deferens) are blocked by mucus and do not develop properly. Men with CBAVD are unable to father children (infertile) unless they undergo fertility treatment. Women with cystic fibrosis may experience complications in pregnancy.
Frequency
Cystic fibrosis is a common genetic disease within the white population in the United States. The disease occurs in 1 in 2,500 to 3,500 white newborns. Cystic fibrosis is less common in other ethnic groups, affecting about 1 in 17,000 African Americans and 1 in 31,000 Asian Americans.
Causes
Mutations in the CFTR gene cause cystic fibrosis. The CFTR gene provides instructions for making a channel that transports negatively charged particles called chloride ions into and out of cells. Chloride is a component of sodium chloride, a common salt found in sweat. Chloride also has important functions in cells; for example, the flow of chloride ions helps control the movement of water in tissues, which is necessary for the production of thin, freely flowing mucus.
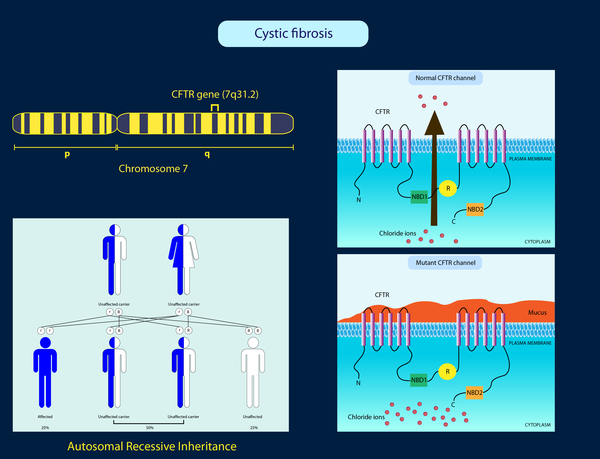
Mutations in the CFTR gene disrupt the function of the chloride channels, preventing them from regulating the flow of chloride ions and water across cell membranes. As a result, cells that line the passageways of the lungs, pancreas, and other organs produce mucus that is unusually thick and sticky. This mucus clogs the airways and various ducts, causing the characteristic signs and symptoms of cystic fibrosis.
Other genetic and environmental factors likely influence the severity of the condition. For example, mutations in genes other than CFTR might help explain why some people with cystic fibrosis are more severely affected than others. Most of these genetic changes have not been identified, however.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- CF
- Cystic fibrosis of pancreas
- Fibrocystic disease of pancreas
- Mucoviscidosis
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Accurso FJ. Update in cystic fibrosis 2005. Am J Respir Crit Care Med. 2006 May 1;173(9):944-7. doi: 10.1164/rccm.2601006. No abstract available. Citation on PubMed or Free article on PubMed Central
- ACOG Committee Opinion No. 486: Update on carrier screening for cystic fibrosis. Obstet Gynecol. 2011 Apr;117(4):1028-1031. doi: 10.1097/AOG.0b013e31821922c2. Citation on PubMed
- Deignan JL, Astbury C, Cutting GR, Del Gaudio D, Gregg AR, Grody WW, Monaghan KG, Richards S; ACMG Laboratory Quality Assurance Committee. CFTR variant testing: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2020 Aug;22(8):1288-1295. doi: 10.1038/s41436-020-0822-5. Epub 2020 May 14. Citation on PubMed
- Gardner J. What you need to know about cystic fibrosis. Nursing. 2007 Jul;37(7):52-5. doi: 10.1097/01.NURSE.0000279437.30155.1e. Citation on PubMed
- Gershman AJ, Mehta AC, Infeld M, Budev MM. Cystic fibrosis in adults: an overview for the internist. Cleve Clin J Med. 2006 Dec;73(12):1065-74. doi: 10.3949/ccjm.73.12.1065. Citation on PubMed
- Grody WW, Thompson BH, Gregg AR, Bean LH, Monaghan KG, Schneider A, Lebo RV. ACMG position statement on prenatal/preconception expanded carrier screening. Genet Med. 2013 Jun;15(6):482-3. doi: 10.1038/gim.2013.47. Epub 2013 Apr 25. Citation on PubMed
- Merlo CA, Boyle MP. Modifier genes in cystic fibrosis lung disease. J Lab Clin Med. 2003 Apr;141(4):237-41. doi: 10.1067/mlc.2003.29. Citation on PubMed
- Ratjen F, Doring G. Cystic fibrosis. Lancet. 2003 Feb 22;361(9358):681-9. doi: 10.1016/S0140-6736(03)12567-6. Citation on PubMed
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005 May 12;352(19):1992-2001. doi: 10.1056/NEJMra043184. No abstract available. Citation on PubMed
- Savant A, Lyman B, Bojanowski C, Upadia J. Cystic Fibrosis. 2001 Mar 26 [updated 2023 Mar 9]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1250/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.