

Description
Cranioectodermal dysplasia is a disorder that affects many parts of the body. The most common features involve bone abnormalities and abnormal development of certain tissues known as ectodermal tissues, which include the skin, hair, nails, and teeth. The signs and symptoms of this condition vary among affected individuals, even among members of the same family.
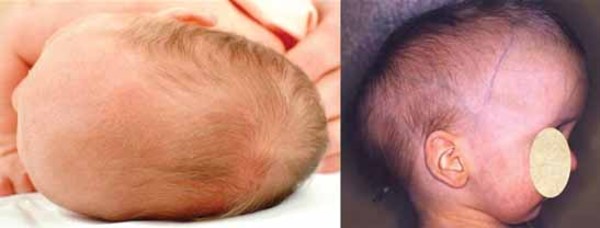
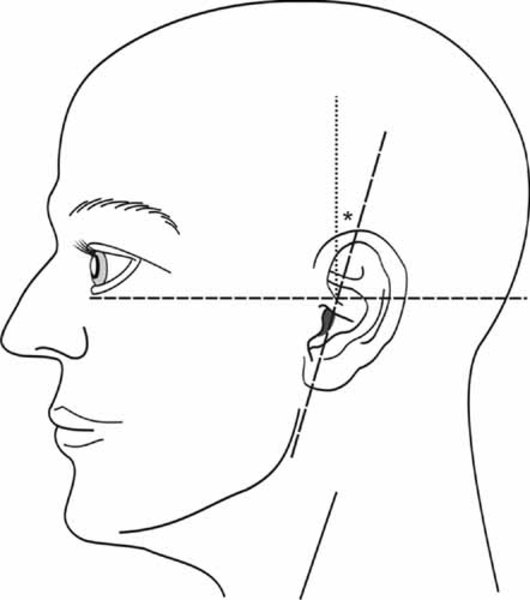
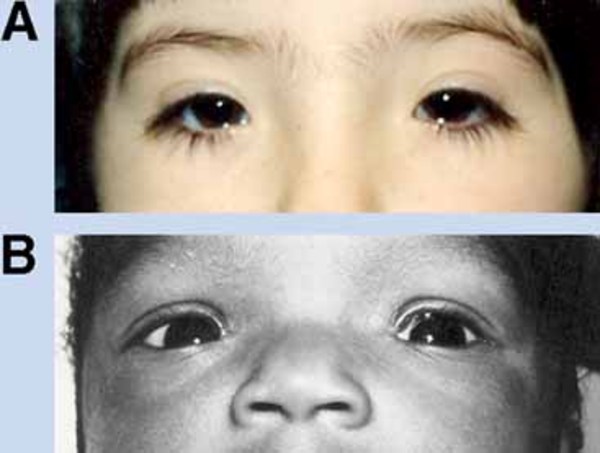
Distinctive abnormalities of the skull and face are common in people with cranioectodermal dysplasia. Most affected individuals have a prominent forehead (frontal bossing) and an elongated head (dolichocephaly) due to abnormal fusion of certain skull bones (sagittal craniosynostosis). A variety of facial abnormalities can occur in people with this condition; these include low-set ears that may also be rotated backward, an increased distance between the inner corners of the eyes (telecanthus), and outside corners of the eyes that point upward or downward (upslanting or downslanting palpebral fissures) among others.
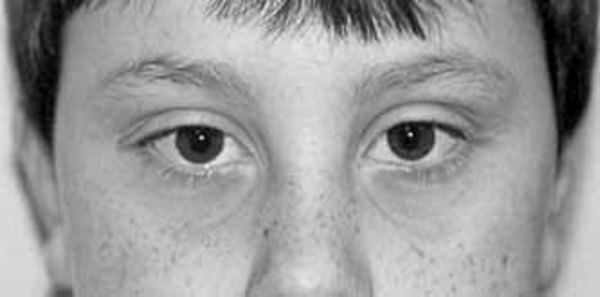
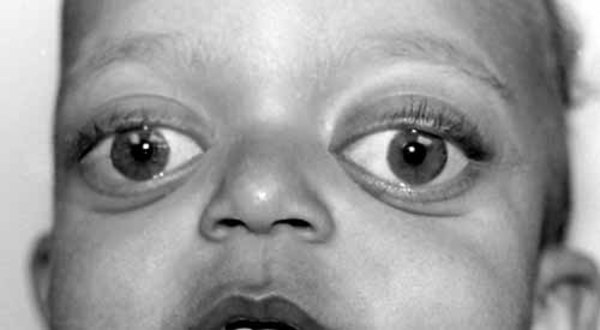
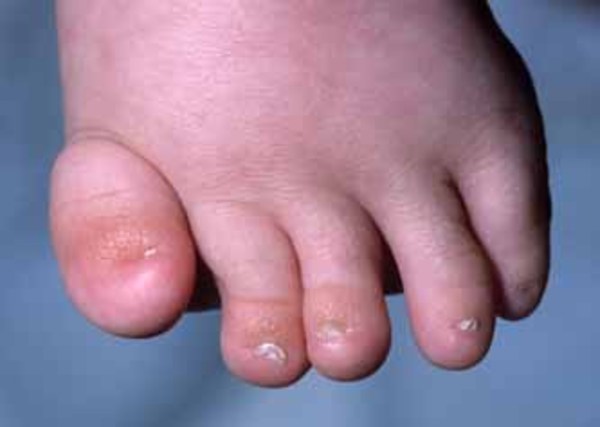
Development of bones in the rest of the skeleton is also affected in this condition. Abnormalities in the long bones of the arms and legs (metaphyseal dysplasia) lead to short limbs and short stature. In addition, affected individuals often have short fingers (brachydactyly). Some people with this condition have short rib bones and a narrow rib cage, which can cause breathing problems, especially in affected newborns.

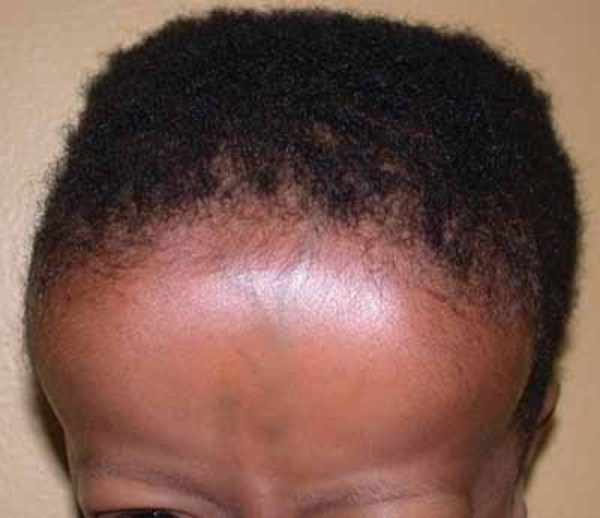
Abnormal development of ectodermal tissues in people with cranioectodermal dysplasia can lead to sparse hair, small or missing teeth, short fingernails and toenails, and loose skin.

Cranioectodermal dysplasia can affect additional organs and tissues in the body. A kidney disorder known as nephronophthisis occurs in many people with this condition, and it can lead to a life-threatening failure of kidney function known as end-stage renal disease. Abnormalities of the liver, heart, or eyes also occur in people with cranioectodermal dysplasia.
Frequency
Cranioectodermal dysplasia is a rare condition with an unknown prevalence. Approximately 40 cases of this condition have been described in the medical literature.
Causes
Cranioectodermal dysplasia is caused by mutations in one of at least four genes: the WDR35, IFT122, WDR19, or IFT43 gene. The protein produced from each of these genes is one piece (subunit) of a protein complex called IFT complex A (IFT-A). This complex is found in finger-like structures called cilia that stick out from the surface of cells. These structures are important for the development and function of many types of cells and tissues. The IFT-A complex is involved in a process called intraflagellar transport, which moves substances within cilia. This movement is essential for the assembly and maintenance of these structures. The IFT-A complex carries materials from the tip to the base of cilia.
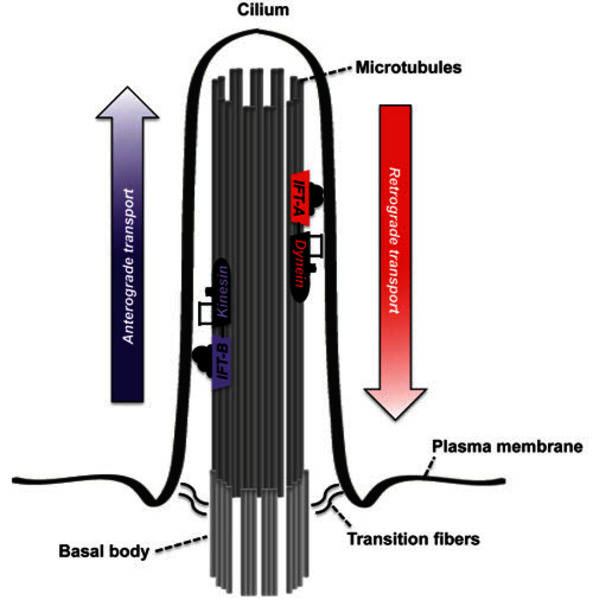
Mutations in any of the four mentioned genes reduce the amount or function of one of the IFT-A subunits. Shortage or abnormal function of a single component of the IFT-A complex impairs the function of the entire complex, disrupting the assembly and maintenance of cilia. These mutations lead to a smaller number of cilia and to abnormalities in their shape and structure. Although the mechanism is unclear, a loss of normal cilia impedes proper development of bone, ectodermal tissues, and other tissues and organs, leading to the features of cranioectodermal dysplasia.
About 40 percent of people with cranioectodermal dysplasia have mutations in one of the four known genes. The cause of the condition in people without mutations in one of these genes is unknown.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- CED
- Sensenbrenner syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Lin AE, Traum AZ, Sahai I, Keppler-Noreuil K, Kukolich MK, Adam MP, Westra SJ, Arts HH. Sensenbrenner syndrome (Cranioectodermal dysplasia): clinical and molecular analyses of 39 patients including two new patients. Am J Med Genet A. 2013 Nov;161A(11):2762-76. doi: 10.1002/ajmg.a.36265. Epub 2013 Oct 3. Citation on PubMed
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002 Nov;3(11):813-25. doi: 10.1038/nrm952. Citation on PubMed
- Tan W, Lin A, Keppler-Noreuil K. Cranioectodermal Dysplasia. 2013 Sep 12 [updated 2022 Dec 15]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK154653/ Citation on PubMed
- Taschner M, Bhogaraju S, Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation. 2012 Feb;83(2):S12-22. doi: 10.1016/j.diff.2011.11.001. Epub 2011 Nov 25. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.