

Description
Congenital nephrotic syndrome is a kidney condition that begins in infancy and typically leads to irreversible kidney failure (end-stage renal disease) by early childhood. Children with congenital nephrotic syndrome begin to have symptoms of the condition between birth and 3 months.
The features of congenital nephrotic syndrome are caused by failure of the kidneys to filter waste products from the blood and remove them in urine. Signs and symptoms of this condition are excessive protein in the urine (proteinuria), increased cholesterol in the blood (hypercholesterolemia), an abnormal buildup of fluid in the abdominal cavity (ascites), and swelling (edema). Affected individuals may also have blood in the urine (hematuria), which can lead to a reduced number of red blood cells (anemia) in the body, abnormal blood clotting, or reduced amounts of certain white blood cells. Low white blood cell counts can lead to a weakened immune system and frequent infections in people with congenital nephrotic syndrome.
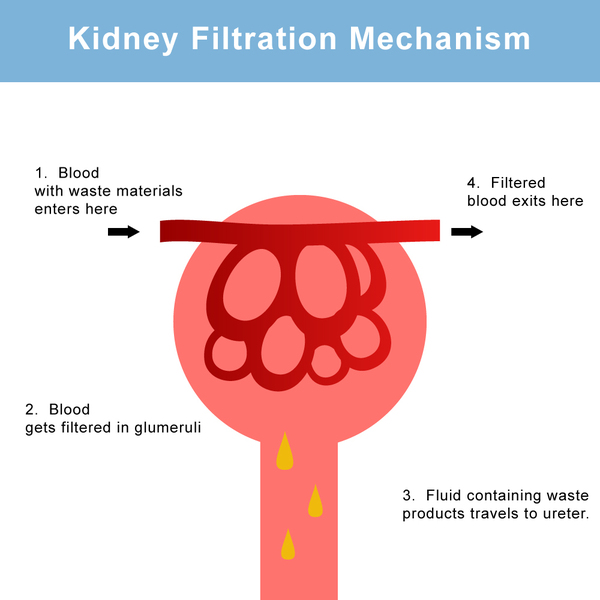

Children with congenital nephrotic syndrome typically develop end-stage renal disease between ages 2 and 8, although with treatment, some may not have kidney failure until adolescence or early adulthood.
Frequency
Congenital nephrotic syndrome affects 1 to 3 per 100,000 children worldwide. In Finland, where this condition is particularly common, congenital nephrotic syndrome is estimated to affect 1 in 10,000 children.
Causes
Mutations in the NPHS1 or NPHS2 gene cause most cases of congenital nephrotic syndrome. These genes provide instructions for making proteins that are found in the kidneys. Specifically, the proteins produced from the NPHS1 and NPHS2 genes are found in cells called podocytes, which are located in specialized kidney structures, called glomeruli, that filter the blood. The proteins are found at the podocyte cell surface in the area between two podocytes called the slit diaphragm. The slit diaphragm is known as a filtration barrier because it captures proteins from blood so that they remain in the body while allowing other molecules like sugars and salts to be excreted in urine. The proteins produced from the NPHS1 and NPHS2 genes also help relay cell signals.

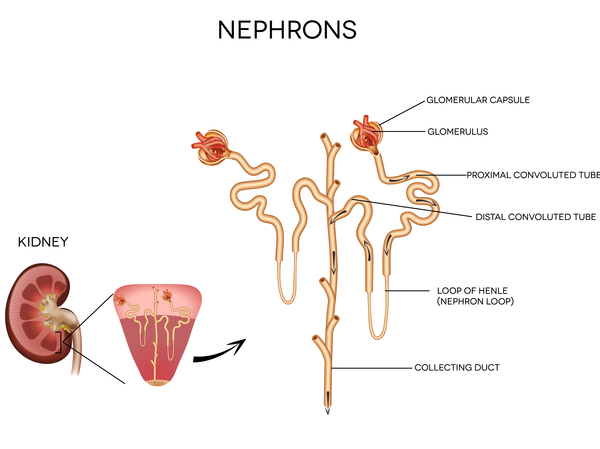
Mutations in the NPHS1 or NPHS2 gene result in a decrease or absence of functional protein, which impairs the formation of normal slit diaphragms. Without a functional slit diaphragm, more molecules pass through the kidneys abnormally and get excreted in urine, including proteins and blood cells. The filtering ability of the kidneys worsens from birth, eventually leading to end-stage renal disease.
NPHS1 gene mutations cause all cases of congenital nephrotic syndrome of the Finnish type. This form of the condition is found in people of Finnish ancestry. NPHS1 gene mutations can cause congenital nephrotic syndrome in non-Finnish individuals, but they are a less common cause than NPHS2 gene mutations, which appear to be the most frequent cause of all cases.
Mutations in other genes cause a small number of cases of congenital nephrotic syndrome. Fifteen to 20 percent of individuals with congenital nephrotic syndrome do not have an identified mutation in one of the genes associated with this condition. In these cases, the cause of the condition may be environmental, including infections such as congenital syphilis or toxoplasmosis, or it may be caused by mutations in unidentified genes.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Familial nephrotic syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD Atlas of Renal Pathology: Congenital Nephrotic Syndrome of Finnish Type. Am J Kidney Dis. 2015 Sep;66(3):e11-2. doi: 10.1053/j.ajkd.2015.07.008. No abstract available. Citation on PubMed
- Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm--from a thin grey line to a complex signalling hub. Nat Rev Nephrol. 2013 Oct;9(10):587-98. doi: 10.1038/nrneph.2013.169. Epub 2013 Sep 3. Citation on PubMed
- Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu J, Hasselbacher K, Hangan D, Ozaltin F, Zenker M, Hildebrandt F; Arbeitsgemeinschaft fur Paediatrische Nephrologie Study Group. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics. 2007 Apr;119(4):e907-19. doi: 10.1542/peds.2006-2164. Epub 2007 Mar 19. Citation on PubMed
- Machuca E, Benoit G, Nevo F, Tete MJ, Gribouval O, Pawtowski A, Brandstrom P, Loirat C, Niaudet P, Gubler MC, Antignac C. Genotype-phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol. 2010 Jul;21(7):1209-17. doi: 10.1681/ASN.2009121309. Epub 2010 May 27. Citation on PubMed or Free article on PubMed Central
- Nishi S, Ubara Y, Utsunomiya Y, Okada K, Obata Y, Kai H, Kiyomoto H, Goto S, Konta T, Sasatomi Y, Sato Y, Nishino T, Tsuruya K, Furuichi K, Hoshino J, Watanabe Y, Kimura K, Matsuo S. Evidence-based clinical practice guidelines for nephrotic syndrome 2014. Clin Exp Nephrol. 2016 Jun;20(3):342-70. doi: 10.1007/s10157-015-1216-x. No abstract available. Citation on PubMed or Free article on PubMed Central
- Ovunc B, Ashraf S, Vega-Warner V, Bockenhauer D, Elshakhs NA, Joseph M, Hildebrandt F; Gesellschaft fur Padiatrische Nephrologie (GPN) Study Group. Mutation analysis of NPHS1 in a worldwide cohort of congenital nephrotic syndrome patients. Nephron Clin Pract. 2012;120(3):c139-46. doi: 10.1159/000337379. Epub 2012 May 11. Citation on PubMed
- Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA; SRNS Study Group; Hildebrandt F. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015 Jun;26(6):1279-89. doi: 10.1681/ASN.2014050489. Epub 2014 Oct 27. Citation on PubMed or Free article on PubMed Central
- Sampson MG, Pollak MR. Opportunities and Challenges of Genotyping Patients With Nephrotic Syndrome in the Genomic Era. Semin Nephrol. 2015 May;35(3):212-21. doi: 10.1016/j.semnephrol.2015.04.002. Citation on PubMed or Free article on PubMed Central
- Schoeb DS, Chernin G, Heeringa SF, Matejas V, Held S, Vega-Warner V, Bockenhauer D, Vlangos CN, Moorani KN, Neuhaus TJ, Kari JA, MacDonald J, Saisawat P, Ashraf S, Ovunc B, Zenker M, Hildebrandt F; Gesselschaft fur Paediatrische Nephrologie (GPN) Study Group. Nineteen novel NPHS1 mutations in a worldwide cohort of patients with congenital nephrotic syndrome (CNS). Nephrol Dial Transplant. 2010 Sep;25(9):2970-6. doi: 10.1093/ndt/gfq088. Epub 2010 Feb 18. Citation on PubMed or Free article on PubMed Central
- Wang JJ, Mao JH. The etiology of congenital nephrotic syndrome: current status and challenges. World J Pediatr. 2016 May;12(2):149-58. doi: 10.1007/s12519-016-0009-y. Epub 2016 Mar 9. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.