

Description
Complete LCAT deficiency is a disorder that primarily affects the eyes and kidneys.
In complete LCAT deficiency, the clear front surface of the eyes (the corneas) gradually becomes cloudy. The cloudiness, which generally first appears in early childhood, consists of small grayish dots of cholesterol (opacities) distributed across the corneas. Cholesterol is a waxy, fat-like substance that is produced in the body and obtained from foods that come from animals; it aids in many functions of the body but can become harmful in excessive amounts. As complete LCAT deficiency progresses, the corneal cloudiness worsens and can lead to severely impaired vision.
People with complete LCAT deficiency often have kidney disease that begins in adolescence or early adulthood. The kidney problems get worse over time and may eventually lead to kidney failure. Individuals with this disorder also usually have a condition known as hemolytic anemia, in which red blood cells are broken down (undergo hemolysis) prematurely, resulting in a shortage of red blood cells (anemia). Anemia can cause pale skin, weakness, fatigue, and more serious complications.


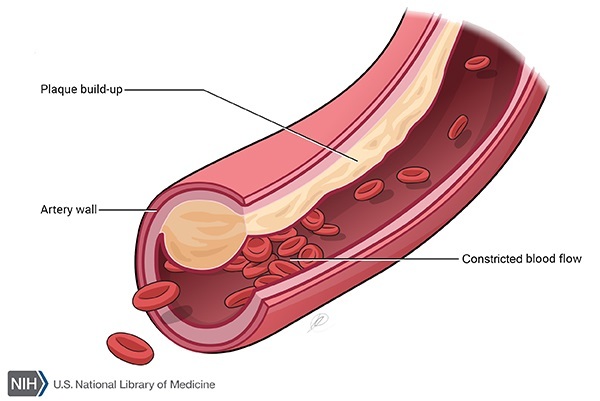
Other features of complete LCAT deficiency that occur in some affected individuals include enlargement of the liver (hepatomegaly), spleen (splenomegaly), or lymph nodes (lymphadenopathy) or an accumulation of fatty deposits on the artery walls (atherosclerosis).

Frequency
Complete LCAT deficiency is a rare disorder. Approximately 70 cases have been reported in the medical literature.
Causes
Complete LCAT deficiency is caused by mutations in the LCAT gene. This gene provides instructions for making an enzyme called lecithin-cholesterol acyltransferase (LCAT).
The LCAT enzyme plays a role in removing cholesterol from the blood and tissues by helping it attach to molecules called lipoproteins, which carry it to the liver. Once in the liver, the cholesterol is redistributed to other tissues or removed from the body. The enzyme has two major functions, called alpha- and beta-LCAT activity. Alpha-LCAT activity helps attach cholesterol to a lipoprotein called high-density lipoprotein (HDL). Beta-LCAT activity helps attach cholesterol to other lipoproteins called very low-density lipoprotein (VLDL) and low-density lipoprotein (LDL).
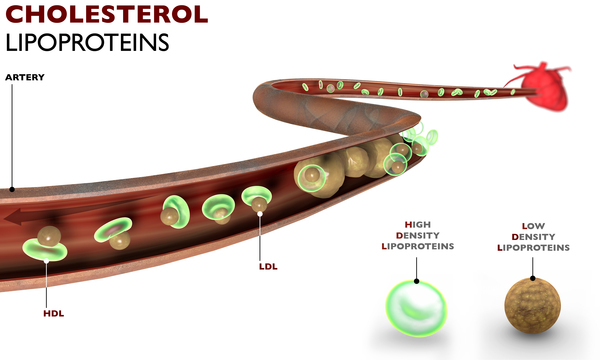
LCAT gene mutations that cause complete LCAT deficiency either prevent the production of LCAT or impair both alpha-LCAT and beta-LCAT activity, reducing the enzyme's ability to attach cholesterol to lipoproteins. Impairment of this mechanism for reducing cholesterol in the body leads to cholesterol deposits in the corneas, kidneys, and other tissues and organs. LCAT gene mutations that affect only alpha-LCAT activity cause a related disorder called fish-eye disease that affects only the corneas.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Familial LCAT deficiency
- Familial lecithin-cholesterol acyltransferase deficiency
- FLD
- LCAT deficiency
- Lecithin acyltransferase deficiency
- Lecithin:cholesterol acyltransferase deficiency
- Norum disease
- Norum's disease
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Calabresi L, Pisciotta L, Costantin A, Frigerio I, Eberini I, Alessandrini P, Arca M, Bon GB, Boscutti G, Busnach G, Frasca G, Gesualdo L, Gigante M, Lupattelli G, Montali A, Pizzolitto S, Rabbone I, Rolleri M, Ruotolo G, Sampietro T, Sessa A, Vaudo G, Cantafora A, Veglia F, Calandra S, Bertolini S, Franceschini G. The molecular basis of lecithin:cholesterol acyltransferase deficiency syndromes: a comprehensive study of molecular and biochemical findings in 13 unrelated Italian families. Arterioscler Thromb Vasc Biol. 2005 Sep;25(9):1972-8. doi: 10.1161/01.ATV.0000175751.30616.13. Epub 2005 Jun 30. Citation on PubMed
- Jahanzad I, Amoueian S, Attaranzadeh A. Familial lecithin-cholesterol acyltransferase deficiency. Arch Iran Med. 2009 Mar;12(2):179-81. Citation on PubMed
- Roshan B, Ganda OP, Desilva R, Ganim RB, Ward E, Haessler SD, Polisecki EY, Asztalos BF, Schaefer EJ. Homozygous lecithin:cholesterol acyltransferase (LCAT) deficiency due to a new loss of function mutation and review of the literature. J Clin Lipidol. 2011 Nov-Dec;5(6):493-9. doi: 10.1016/j.jacl.2011.07.002. Epub 2011 Aug 23. Citation on PubMed or Free article on PubMed Central
- Savel J, Lafitte M, Pucheu Y, Pradeau V, Tabarin A, Couffinhal T. Very low levels of HDL cholesterol and atherosclerosis, a variable relationship--a review of LCAT deficiency. Vasc Health Risk Manag. 2012;8:357-61. doi: 10.2147/VHRM.S29985. Epub 2012 Jun 5. Citation on PubMed or Free article on PubMed Central
- Shoji K, Morita H, Ishigaki Y, Rivard CJ, Takayasu M, Nakayama K, Nakayama T, Inoue Y, Ayaki M, Yoshimura A. Lecithin-cholesterol acyltransferase (LCAT) deficiency without mutations in the coding sequence: a case report and literature review. Clin Nephrol. 2011 Oct;76(4):323-8. doi: 10.5414/cn106484. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.