

Description
Combined oxidative phosphorylation deficiency 1 is a severe condition that primarily impairs neurological and liver function.
Most people with combined oxidative phosphorylation deficiency 1 have severe brain dysfunction (encephalopathy) that worsens over time; they also have difficulty growing and gaining weight at the expected rate (failure to thrive). In some cases, affected individuals have abnormal muscle tone (increased or decreased), developmental delay, seizures, loss of sensation in the limbs (peripheral neuropathy), and an unusually small head (microcephaly). Liver disease is common in people with combined oxidative phosphorylation deficiency 1, with individuals quickly developing liver failure. Individuals with this condition also usually have a potentially life-threatening buildup of a chemical called lactic acid in the body (lactic acidosis).

The neurological features of combined oxidative phosphorylation deficiency 1 are largely due to brain abnormalities that include thinning of the tissue that connects the two halves of the brain (corpus callosum hypoplasia) and loss of brain tissue called white matter (leukodystrophy), particularly in an area of the brain called the basal ganglia, which normally helps control movement.

Individuals with combined oxidative phosphorylation deficiency 1 usually do not survive past early childhood, although some people live longer.
Frequency
Combined oxidative phosphorylation deficiency 1 is likely a rare disorder, although its prevalence is unknown. At least 12 affected individuals have been described in the scientific literature.
Causes
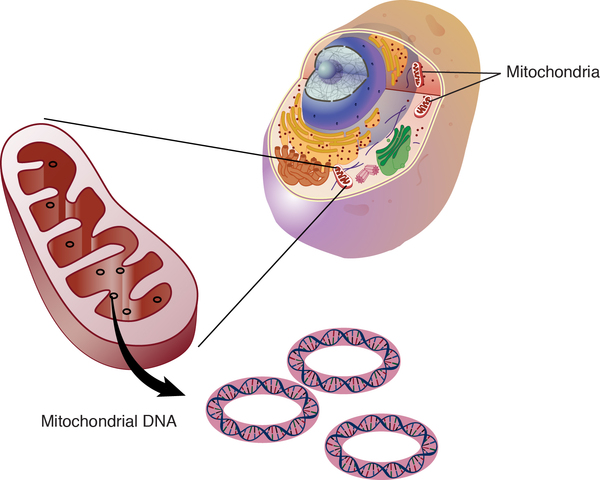
Combined oxidative phosphorylation deficiency 1 is caused by mutations in the GFM1 gene. This gene provides instructions for making an enzyme called mitochondrial translation elongation factor G1. This enzyme is found in cell structures called mitochondria, which are the energy-producing centers in cells.

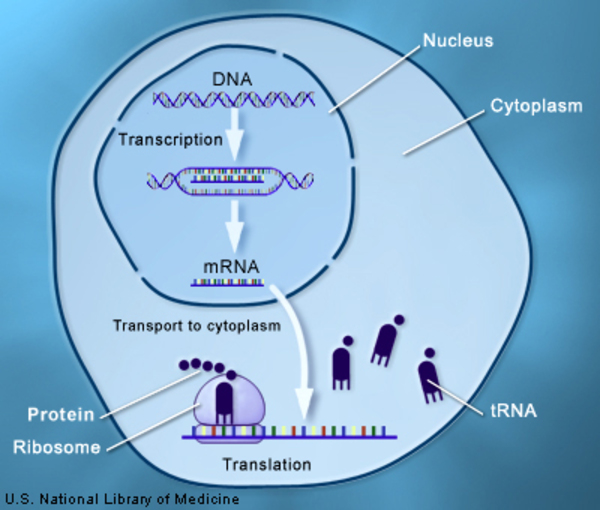
While instructions for making most of the body's proteins are found in DNA that is stored in the nucleus of cells (nuclear DNA), a few proteins and other molecules are produced from DNA that is stored in mitochondria (mtDNA). Mitochondrial translation elongation factor G1 is involved in the production of proteins from mtDNA genes through a process called translation. The enzyme's role in translation is to coordinate the movements of mtRNA molecules, which are the protein blueprints created from mtDNA. This function allows assembly of proteins to continue until it is complete. Genes on mtDNA provide instructions for proteins that are primarily involved in the process of converting the energy from food into a form cells can use (oxidative phosphorylation).
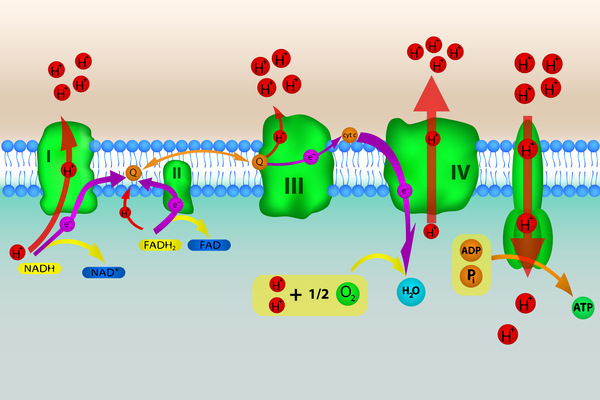
GFM1 gene mutations reduce or eliminate mitochondrial translation elongation factor G1 function. As a result, fewer mitochondrial proteins involved in oxidative phosphorylation are produced. (The process of oxidative phosphorylation involves five groups of proteins, or complexes. The condition is called combined oxidative phosphorylation deficiency 1 because it impairs the function of more than one of these complexes.) Organs that have high energy demands, such as the brain and liver, are particularly affected by the resulting impairment of oxidative phosphorylation. A shortage of energy in these tissues leads to cell death, causing the neurological and liver problems in people with combined oxidative phosphorylation deficiency 1. It is thought that other tissues that require a lot of energy, such as the heart and other muscles, are not affected in this condition because they have additional enzymes that can perform the process of mitochondrial protein production.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- COXPD1
- Early fatal progressive hepatoencephalopathy
- Hepatoencephalopathy due to combined oxidative phosphorylation defect type 1
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet. 2006 Jun 1;15(11):1835-46. doi: 10.1093/hmg/ddl106. Epub 2006 Apr 21. Citation on PubMed
- Balasubramaniam S, Choy YS, Talib A, Norsiah MD, van den Heuvel LP, Rodenburg RJ. Infantile Progressive Hepatoencephalomyopathy with Combined OXPHOS Deficiency due to Mutations in the Mitochondrial Translation Elongation Factor Gene GFM1. JIMD Rep. 2012;5:113-22. doi: 10.1007/8904_2011_107. Epub 2011 Dec 21. Citation on PubMed or Free article on PubMed Central
- Brito S, Thompson K, Campistol J, Colomer J, Hardy SA, He L, Fernandez-Marmiesse A, Palacios L, Jou C, Jimenez-Mallebrera C, Armstrong J, Montero R, Artuch R, Tischner C, Wenz T, McFarland R, Taylor RW. Long-term survival in a child with severe encephalopathy, multiple respiratory chain deficiency and GFM1 mutations. Front Genet. 2015 Mar 23;6:102. doi: 10.3389/fgene.2015.00102. eCollection 2015. Erratum In: Front Genet. 2015;6:254. Citation on PubMed or Free article on PubMed Central
- Coenen MJ, Antonicka H, Ugalde C, Sasarman F, Rossi R, Heister JG, Newbold RF, Trijbels FJ, van den Heuvel LP, Shoubridge EA, Smeitink JA. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med. 2004 Nov 11;351(20):2080-6. doi: 10.1056/NEJMoa041878. Citation on PubMed
- Ravn K, Schonewolf-Greulich B, Hansen RM, Bohr AH, Duno M, Wibrand F, Ostergaard E. Neonatal mitochondrial hepatoencephalopathy caused by novel GFM1 mutations. Mol Genet Metab Rep. 2015 Feb 20;3:5-10. doi: 10.1016/j.ymgmr.2015.01.004. eCollection 2015 Jun. Citation on PubMed or Free article on PubMed Central
- Smits P, Antonicka H, van Hasselt PM, Weraarpachai W, Haller W, Schreurs M, Venselaar H, Rodenburg RJ, Smeitink JA, van den Heuvel LP. Mutation in subdomain G' of mitochondrial elongation factor G1 is associated with combined OXPHOS deficiency in fibroblasts but not in muscle. Eur J Hum Genet. 2011 Mar;19(3):275-9. doi: 10.1038/ejhg.2010.208. Epub 2010 Dec 1. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.