

Description
CLN4 disease is a condition that primarily affects the nervous system, causing problems with movement and intellectual function that worsen over time. The signs and symptoms of CLN4 disease typically appear around age 30, but they can develop anytime between adolescence and late adulthood.
People with CLN4 disease often develop seizures and uncontrollable muscle jerks (myoclonic epilepsy), a decline in intellectual function (dementia), problems with coordination and balance (ataxia), tremors or other involuntary movements (motor tics), and speech difficulties (dysarthria). The signs and symptoms of CLN4 disease worsen over time, and affected individuals usually survive about 15 years after the disorder begins.
CLN4 disease is one of a group of disorders known as neuronal ceroid lipofuscinoses (NCLs), which may also be collectively referred to as Batten disease. (The adult forms of NCLs, which includes CLN4 disease, are sometimes known as Kufs disease.) All the NCLs affect the nervous system and typically cause worsening problems with vision, movement, and thinking ability. The different NCLs are distinguished by their genetic cause. Each disease type is given the designation "CLN," meaning ceroid lipofuscinosis, neuronal, and then a number to indicate its subtype.
Frequency
CLN4 disease is a rare disorder, but its prevalence is unknown. Collectively, all forms of NCL affect an estimated 1 in 100,000 individuals worldwide.
Causes
Mutations in the DNAJC5 gene cause CLN4 disease. The DNAJC5 gene provides instructions for making a protein called cysteine string protein alpha (CSPα). This protein is found in the brain, where it plays a role in the transmission of nerve impulses, helping nerve cells communicate with each other. Specifically, CSPα is involved in recycling certain proteins that are involved in nerve impulse transmission by refolding misshapen proteins so that they can be used in additional transmissions.
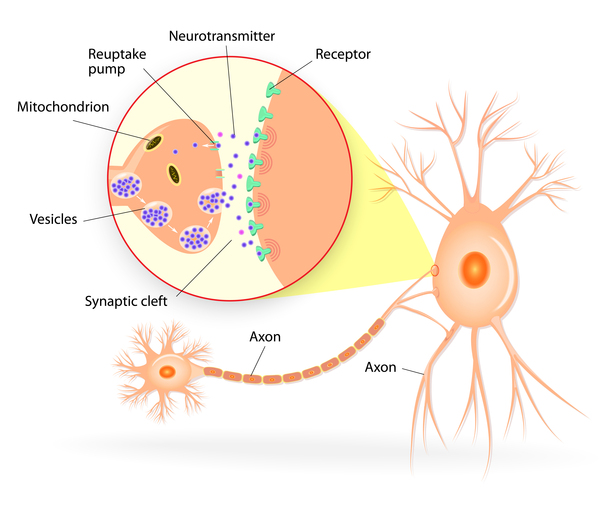
DNAJC5 gene mutations lead to the production of an altered CSPα protein. The altered protein cannot perform its function, which reduces protein recycling, causing a shortage (deficiency) of functional proteins needed for impulse transmission. Without normal communication between nerve cells, neurological functions are impaired, contributing to the features of CLN4 disease.
CLN4 disease, like other NCLs, is characterized by the accumulation of proteins and other substances in lysosomes, which are compartments in the cell that digest and recycle materials. These accumulations occur in cells throughout the body; however, nerve cells seem to be particularly vulnerable to their effects. The accumulations can cause cell damage leading to cell death. The progressive death of nerve cells in the brain and other tissues contributes to the decline of neurological function in CLN4 disease. However, it is unclear how mutations in the DNAJC5 gene are involved in the buildup of substances in lysosomes.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Some cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual’s parent or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- Adult neuronal ceroid lipofuscinosis
- Ceroid lipofuscinosis, neuronal, 4B, autosomal dominant
- CLN4B
- Parry disease
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Cadieux-Dion M, Andermann E, Lachance-Touchette P, Ansorge O, Meloche C, Barnabe A, Kuzniecky RI, Andermann F, Faught E, Leonberg S, Damiano JA, Berkovic SF, Rouleau GA, Cossette P. Recurrent mutations in DNAJC5 cause autosomal dominant Kufs disease. Clin Genet. 2013 Jun;83(6):571-5. doi: 10.1111/cge.12020. Epub 2012 Nov 7. Citation on PubMed
- Henderson MX, Wirak GS, Zhang YQ, Dai F, Ginsberg SD, Dolzhanskaya N, Staropoli JF, Nijssen PC, Lam TT, Roth AF, Davis NG, Dawson G, Velinov M, Chandra SS. Neuronal ceroid lipofuscinosis with DNAJC5/CSPalpha mutation has PPT1 pathology and exhibit aberrant protein palmitoylation. Acta Neuropathol. 2016 Apr;131(4):621-37. doi: 10.1007/s00401-015-1512-2. Epub 2015 Dec 10. Citation on PubMed or Free article on PubMed Central
- Kollmann K, Uusi-Rauva K, Scifo E, Tyynela J, Jalanko A, Braulke T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim Biophys Acta. 2013 Nov;1832(11):1866-81. doi: 10.1016/j.bbadis.2013.01.019. Epub 2013 Feb 9. Citation on PubMed
- Schulz A, Kohlschutter A, Mink J, Simonati A, Williams R. NCL diseases - clinical perspectives. Biochim Biophys Acta. 2013 Nov;1832(11):1801-6. doi: 10.1016/j.bbadis.2013.04.008. Epub 2013 Apr 17. Citation on PubMed or Free article on PubMed Central
- Smith KR, Dahl HH, Canafoglia L, Andermann E, Damiano J, Morbin M, Bruni AC, Giaccone G, Cossette P, Saftig P, Grotzinger J, Schwake M, Andermann F, Staropoli JF, Sims KB, Mole SE, Franceschetti S, Alexander NA, Cooper JD, Chapman HA, Carpenter S, Berkovic SF, Bahlo M. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum Mol Genet. 2013 Apr 1;22(7):1417-23. doi: 10.1093/hmg/dds558. Epub 2013 Jan 7. Citation on PubMed or Free article on PubMed Central
- Velinov M, Dolzhanskaya N, Gonzalez M, Powell E, Konidari I, Hulme W, Staropoli JF, Xin W, Wen GY, Barone R, Coppel SH, Sims K, Brown WT, Zuchner S. Mutations in the gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PLoS One. 2012;7(1):e29729. doi: 10.1371/journal.pone.0029729. Epub 2012 Jan 3. Erratum In: PLoS One. 2012;7(9). doi:10.1371/annotation/26d7eb64-ccd2-41db-b1aa-7cdc8c1eff95. Citation on PubMed or Free article on PubMed Central
- Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012 Jul 10;79(2):183-91. doi: 10.1212/WNL.0b013e31825f0547. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.