

Description
Charcot-Marie-Tooth disease encompasses a group of disorders called hereditary sensory and motor neuropathies that damage the peripheral nerves. Peripheral nerves connect the brain and spinal cord to muscles and to sensory cells that detect sensations such as touch, pain, heat, and sound. Damage to the peripheral nerves that worsens over time can result in alteration or loss of sensation and wasting (atrophy) of muscles in the feet, legs, and hands.
Charcot-Marie-Tooth disease usually becomes apparent in adolescence or early adulthood, but onset may occur anytime from early childhood through late adulthood. Symptoms of Charcot-Marie-Tooth disease vary in severity and age of onset even among members of the same family. Some people never realize they have the disorder because their symptoms are so mild, but most have a moderate amount of physical disability. A small percentage of people experience severe weakness or other problems which, in very rare cases, can be life-threatening. In most affected individuals, however, Charcot-Marie-Tooth disease does not affect life expectancy.
Typically, the earliest symptoms of Charcot-Marie-Tooth disease result from muscle atrophy in the feet. Affected individuals may have foot abnormalities such as high arches (pes cavus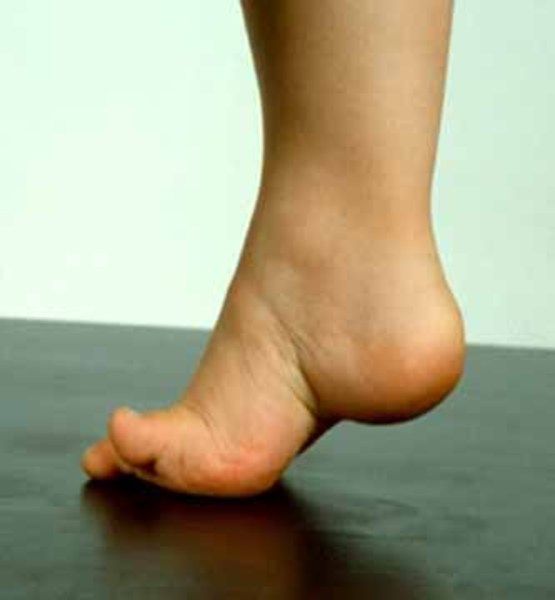 ), flat feet (pes planus
), flat feet (pes planus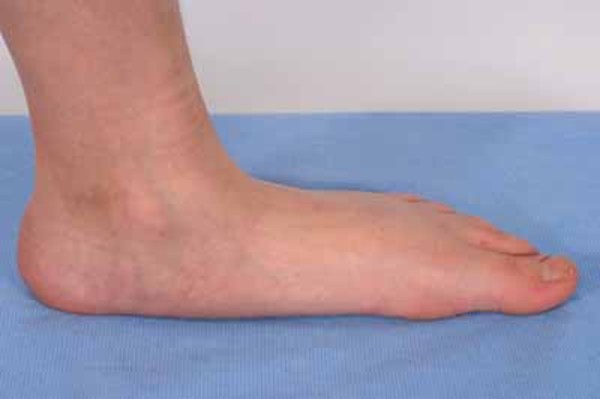 ), or curled toes (hammer toes). They often have difficulty flexing the foot or walking on the heel of the foot. These difficulties may cause a higher than normal step (steppage gait) and increase the risk of ankle injuries and tripping. As the disease worsens, muscles in the lower legs usually weaken, but leg and foot problems rarely require the use of a wheelchair.
), or curled toes (hammer toes). They often have difficulty flexing the foot or walking on the heel of the foot. These difficulties may cause a higher than normal step (steppage gait) and increase the risk of ankle injuries and tripping. As the disease worsens, muscles in the lower legs usually weaken, but leg and foot problems rarely require the use of a wheelchair.
Affected individuals may also develop weakness in the hands, causing difficulty with daily activities such as writing, fastening buttons, and turning doorknobs. People with Charcot-Marie-Tooth disease typically experience a decreased sensitivity to touch, heat, and cold in the feet and lower legs, but occasionally feel aching or burning sensations. In rare cases, affected individuals have loss of vision or gradual hearing loss that sometimes leads to deafness.
There are several types of Charcot-Marie-Tooth disease, which are differentiated by their effects on nerve cells and patterns of inheritance. Type 1 (CMT1) is characterized by abnormalities in myelin, the fatty substance that covers nerve cells, protecting them and helping to transmit nerve impulses. These abnormalities slow the transmission of nerve impulses and can affect the health of the nerve fiber. Type 2 (CMT2) is characterized by abnormalities in the fiber, or axon , that extends from a nerve cell body to muscles or to sense organs. These abnormalities reduce the strength of the nerve impulse. People with CMT2 may develop amyotrophic lateral sclerosis (ALS), a condition characterized by progressive muscle weakness, a loss of muscle mass, and an inability to control movement.
In forms of Charcot-Marie-Tooth disease classified as intermediate type, the nerve impulses are both slowed and reduced in strength, probably due to abnormalities in both myelin and axons. Type 4 (CMT4) is distinguished from the other types by its pattern of inheritance; it can affect either the axons or the myelin. Type X Charcot-Marie-Tooth disease (CMTX) is caused by mutations in genes on the X chromosome, one of the two sex chromosomes. Within the various types of Charcot-Marie-Tooth disease, subtypes (such as CMT1A, CMT1B, CMT2A, CMT4A, and CMTX1) indicate different genetic causes.
, that extends from a nerve cell body to muscles or to sense organs. These abnormalities reduce the strength of the nerve impulse. People with CMT2 may develop amyotrophic lateral sclerosis (ALS), a condition characterized by progressive muscle weakness, a loss of muscle mass, and an inability to control movement.
In forms of Charcot-Marie-Tooth disease classified as intermediate type, the nerve impulses are both slowed and reduced in strength, probably due to abnormalities in both myelin and axons. Type 4 (CMT4) is distinguished from the other types by its pattern of inheritance; it can affect either the axons or the myelin. Type X Charcot-Marie-Tooth disease (CMTX) is caused by mutations in genes on the X chromosome, one of the two sex chromosomes. Within the various types of Charcot-Marie-Tooth disease, subtypes (such as CMT1A, CMT1B, CMT2A, CMT4A, and CMTX1) indicate different genetic causes.
Sometimes other, historical names are used to refer to particular forms of Charcot-Marie-Tooth disease. For example, Roussy-Levy syndrome is a form of CMT11 with the additional feature of rhythmic shaking (tremors). Dejerine-Sottas syndrome is a term sometimes used to describe a severe, early childhood form of Charcot-Marie-Tooth disease; it is also sometimes called type 3 (CMT3). Depending on the specific gene that is altered, this severe, early-onset form of the disorder may also be classified as CMT1 or CMT4. CMTX5 is also known as Rosenberg-Chutorian syndrome.
Frequency
Charcot-Marie-Tooth disease is the most common inherited disorder that involves the peripheral nerves, affecting an estimated 150,000 people in the United States. It occurs in populations worldwide with a prevalence of about 1 in 3,300 individuals.
Causes
Charcot-Marie-Tooth disease can be caused by mutations in many different genes. These genes provide instructions for making proteins that are involved in the function of peripheral nerves in the feet, legs, and hands. The gene mutations that cause Charcot-Marie-Tooth disease affect the function of the proteins in ways that are not fully understood; however, they likely impair axons, which transmit nerve impulses, or affect the specialized cells that produce myelin. In most cases, longer nerves that transmit impulses to the appendages of the body are more likely to be affected. As a result, peripheral nerve cells slowly lose the ability to stimulate the muscles in the feet, legs, and eventually the hands, and to transmit sensory signals from these appendages to the brain. Different mutations within a single gene may cause signs and symptoms of differing severities or lead to different types of Charcot-Marie-Tooth disease.
Between 70 and 80 percent of individuals with CMT1 have mutations affecting the PMP22 gene. Most of these cases occur when there is an extra copy of the gene resulting from a small duplication of genetic material on chromosome 17. Another 10 to 12 percent of individuals with CMT1 have mutations in the MPZ gene. MPZ gene mutations are also occasionally identified in people with other forms of the disorder. The most common cause of CMT2 is mutations in the MFN2 gene, which accounts for about 20 percent of cases. Approximately 90 percent of people with CMTX have GJB1 gene mutations. Mutations in dozens of other genes have been identified in smaller numbers of people with these and the other types. The list of genes associated with Charcot-Marie-Tooth disease continues to grow as researchers study this disorder.
Inheritance
The pattern of inheritance varies with the type of Charcot-Marie-Tooth disease. CMT1, most cases of CMT2, and most intermediate forms are inherited in an autosomal dominant pattern. This pattern of inheritance means that one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one affected parent. Each of the children of an affected parent has a 50 percent chance of inheriting the disorder.

CMT4, a few CMT2 subtypes, and some intermediate forms are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition. Children of affected individuals are not affected unless the other parent also passes down a mutation in the same gene.

CMTX is inherited in an X-linked dominant pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome. The inheritance is dominant if one copy of the altered gene is sufficient to cause the condition. In most cases, affected males, who have the alteration on their only copy of the X chromosome, experience more severe symptoms of the disorder than affected females, who have the alteration on one of their two X chromosomes. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons. All daughters of affected men will have one altered X chromosome, but they may have only mild symptoms of the disorder.
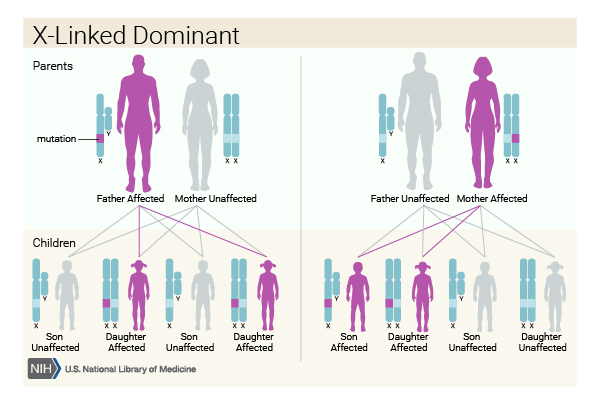
Some cases of autosomal dominant or type X Charcot-Marie-Tooth disease result from a new mutation and occur in people with no history of the disorder in their family.
Other Names for This Condition
- Charcot-Marie-Tooth hereditary neuropathy
- Charcot-Marie-Tooth syndrome
- CMT
- Hereditary motor and sensory neuropathy
- HMSN
- Peroneal muscular atrophy
- PMA
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1B; CMT1B
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2A1; CMT2A1
- CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1A; CMT1A
- CHARCOT-MARIE-TOOTH DISEASE, GUADALAJARA NEURONAL TYPE
- CHARCOT-MARIE-TOOTH DISEASE AND DEAFNESS
- KERATODERMA, PALMOPLANTAR, WITH NAIL DYSTROPHY AND HEREDITARY MOTOR-SENSORY NEUROPATHY
- HYPERTROPHIC NEUROPATHY OF DEJERINE-SOTTAS
- CHARCOT-MARIE-TOOTH DISEASE, TYPE 4D; CMT4D
- HEREDITARY MOTOR AND SENSORY NEUROPATHY V
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2B; CMT2B
- CHARCOT-MARIE-TOOTH DISEASE, TYPE 4B1; CMT4B1
- CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1C; CMT1C
- NEUROPATHY, HEREDITARY MOTOR AND SENSORY, WITH EXCESSIVE MYELIN FOLDING COMPLEX, AUTOSOMAL RECESSIVE
- NEUROPATHY, HEREDITARY MOTOR AND SENSORY, WITH DEAFNESS, IMPAIRED INTELLECTUAL DEVELOPMENT, AND ABSENT SENSORY LARGE MYELINATED FIBERS
- CHARCOT-MARIE-TOOTH DISEASE, TYPE 4A; CMT4A
- CHARCOT-MARIE-TOOTH DISEASE, X-LINKED RECESSIVE, 4, WITH OR WITHOUT CEREBELLAR ATAXIA; CMTX4
- CHARCOT-MARIE-TOOTH DISEASE, X-LINKED RECESSIVE, 5; CMTX5
- ROUSSY-LEVY HEREDITARY AREFLEXIC DYSTASIA
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2D; CMT2D
- CHARCOT-MARIE-TOOTH DISEASE, TYPE 4C; CMT4C
- CHARCOT-MARIE-TOOTH DISEASE, X-LINKED DOMINANT, 1; CMTX1
- NEUROPATHY, HEREDITARY MOTOR AND SENSORY, TYPE VIA, WITH OPTIC ATROPHY; HMSN6A
- NEUROPATHY, HEREDITARY MOTOR AND SENSORY, RUSSE TYPE; HMSNR
- NEUROPATHY, CONGENITAL HYPOMYELINATING, 1, AUTOSOMAL RECESSIVE; CHN1
- CHARCOT-MARIE-TOOTH DISEASE, TYPE 4B2; CMT4B2
- CHARCOT-MARIE-TOOTH DISEASE, DOMINANT INTERMEDIATE D; CMTDID
- CHARCOT-MARIE-TOOTH DISEASE, DOMINANT INTERMEDIATE C; CMTDIC
- CHARCOT-MARIE-TOOTH DISEASE, RECESSIVE INTERMEDIATE A; CMTRIA
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2L; CMT2L
- HEREDITARY MOTOR AND SENSORY NEUROPATHY, TYPE IIC; HMSN2C
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2K; CMT2K
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, WITH VOCAL CORD PARESIS, AUTOSOMAL RECESSIVE
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2I; CMT2I
- CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1D; CMT1D
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2E; CMT2E
- CHARCOT-MARIE-TOOTH DISEASE, DOMINANT INTERMEDIATE B; CMTDIB
- CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1F; CMT1F
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2J; CMT2J
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2B1; CMT2B1
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2B2; CMT2B2
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2F; CMT2F
- CHARCOT-MARIE-TOOTH DISEASE, TYPE 4J; CMT4J
- CHARCOT-MARIE-TOOTH DISEASE, TYPE 4H; CMT4H
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, AUTOSOMAL DOMINANT, TYPE 2A2A; CMT2A2A
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2O; CMT2O
- CHARCOT-MARIE-TOOTH DISEASE, DOMINANT INTERMEDIATE E; CMTDIE
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2N; CMT2N
- NEUROPATHY, HEREDITARY MOTOR AND SENSORY, TYPE VIB, WITH OPTIC ATROPHY; HMSN6B
- CHARCOT-MARIE-TOOTH DISEASE, AXONAL, TYPE 2P; CMT2P
Scientific Articles on PubMed
References
- Ando M, Higuchi Y, Yuan J, Yoshimura A, Taniguchi T, Takei J, Takeuchi M, Hiramatsu Y, Shimizu F, Kubota M, Takeshima A, Ueda T, Koh K, Nagaoka U, Tokashiki T, Sawai S, Sakiyama Y, Hashiguchi A, Sato R, Kanda T, Okamoto Y, Takashima H. Novel heterozygous variants of SLC12A6 in Japanese families with Charcot-Marie-Tooth disease. Ann Clin Transl Neurol. 2022 Jul;9(7):902-911. doi: 10.1002/acn3.51603. Epub 2022 Jun 22. Citation on PubMed
- Barreto LC, Oliveira FS, Nunes PS, de Franca Costa IM, Garcez CA, Goes GM, Neves EL, de Souza Siqueira Quintans J, de Souza Araujo AA. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology. 2016;46(3):157-65. doi: 10.1159/000443706. Epub 2016 Feb 6. Citation on PubMed
- Berciano J, Garcia A, Gallardo E, Peeters K, Pelayo-Negro AL, Alvarez-Paradelo S, Gazulla J, Martinez-Tames M, Infante J, Jordanova A. Intermediate Charcot-Marie-Tooth disease: an electrophysiological reappraisal and systematic review. J Neurol. 2017 Aug;264(8):1655-1677. doi: 10.1007/s00415-017-8474-3. Epub 2017 Mar 31. Citation on PubMed
- Bird TD. Charcot-Marie-Tooth Hereditary Neuropathy Overview. 1998 Sep 28 [updated 2024 Mar 14]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1358/ Citation on PubMed
- Echaniz-Laguna A. The shifting paradigm of Charcot-Marie-Tooth disease. Rev Neurol (Paris). 2015 Jun-Jul;171(6-7):498-504. doi: 10.1016/j.neurol.2014.12.003. Epub 2015 Apr 18. Citation on PubMed
- Ekins S, Litterman NK, Arnold RJ, Burgess RW, Freundlich JS, Gray SJ, Higgins JJ, Langley B, Willis DE, Notterpek L, Pleasure D, Sereda MW, Moore A. A brief review of recent Charcot-Marie-Tooth research and priorities. F1000Res. 2015 Feb 26;4:53. doi: 10.12688/f1000research.6160.1. eCollection 2015. Citation on PubMed or Free article on PubMed Central
- El-Abassi R, England JD, Carter GT. Charcot-Marie-Tooth disease: an overview of genotypes, phenotypes, and clinical management strategies. PM R. 2014 Apr;6(4):342-55. doi: 10.1016/j.pmrj.2013.08.611. Epub 2014 Jan 13. Citation on PubMed
- Gutmann L, Shy M. Update on Charcot-Marie-Tooth disease. Curr Opin Neurol. 2015 Oct;28(5):462-7. doi: 10.1097/WCO.0000000000000237. Citation on PubMed
- Hoyle JC, Isfort MC, Roggenbuck J, Arnold WD. The genetics of Charcot-Marie-Tooth disease: current trends and future implications for diagnosis and management. Appl Clin Genet. 2015 Oct 19;8:235-43. doi: 10.2147/TACG.S69969. eCollection 2015. Citation on PubMed or Free article on PubMed Central
- Jani-Acsadi A, Ounpuu S, Pierz K, Acsadi G. Pediatric Charcot-Marie-Tooth disease. Pediatr Clin North Am. 2015 Jun;62(3):767-86. doi: 10.1016/j.pcl.2015.03.012. Epub 2015 Apr 15. Citation on PubMed
- Kazamel M, Boes CJ. Charcot Marie Tooth disease (CMT): historical perspectives and evolution. J Neurol. 2015;262(4):801-5. doi: 10.1007/s00415-014-7490-9. Epub 2014 Sep 9. Citation on PubMed
- Kim JW, Kim HJ. Charcot-Marie-Tooth Neuropathy X Type 5 - RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. 2008 Aug 26 [updated 2013 Jun 6]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1876/ Citation on PubMed
- Wang Y, Yin F. A Review of X-linked Charcot-Marie-Tooth Disease. J Child Neurol. 2016 May;31(6):761-72. doi: 10.1177/0883073815604227. Epub 2015 Sep 18. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.