

Description
CDKL5 deficiency disorder is characterized by seizures that begin in infancy, followed by significant delays in many aspects of development.
Seizures in CDKL5 deficiency disorder usually begin within the first 3 months of life, and can appear as early as the first week after birth. The types of seizures change with age, and may follow a predictable pattern. The most common types are generalized tonic-clonic seizures, which involve a loss of consciousness, muscle rigidity, and convulsions; tonic seizures, which are characterized by abnormal muscle contractions; and epileptic spasms, which involve short episodes of muscle jerks. Seizures occur daily in most people with CDKL5 deficiency disorder, although they can have periods when they are seizure-free. Seizures in CDKL5 deficiency disorder usually do not get better with treatment.
Development is impaired in children with CDKL5 deficiency disorder. Most have severe intellectual disability and little or no speech. The development of gross motor skills, such as sitting, standing, and walking, is delayed or not achieved. About one-third of affected individuals are able to walk independently. Fine motor skills, such as picking up small objects with the fingers, are also impaired; about half of affected individuals have purposeful use of their hands. Most people with this condition have vision problems (cortical visual impairment).
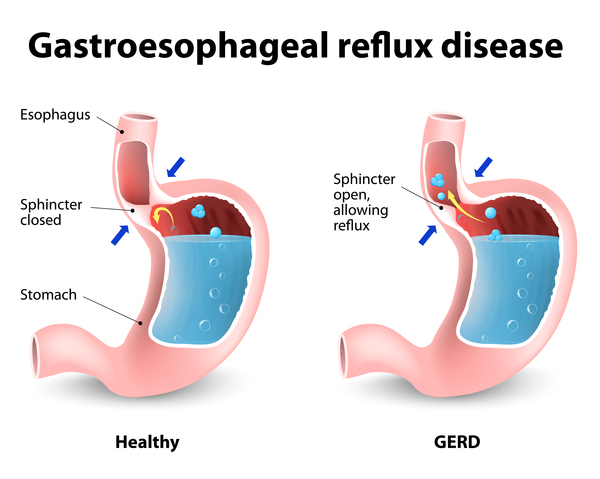
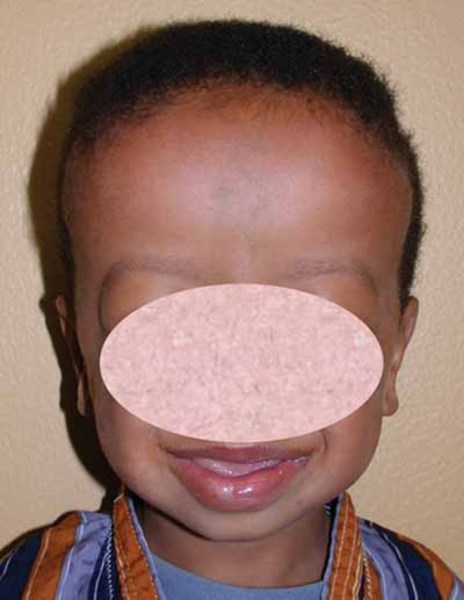
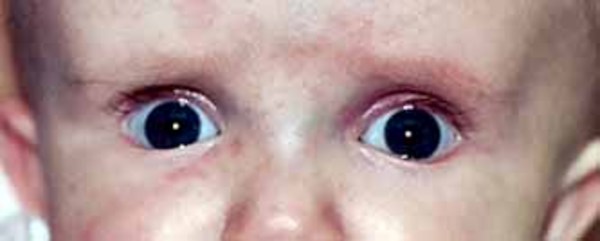
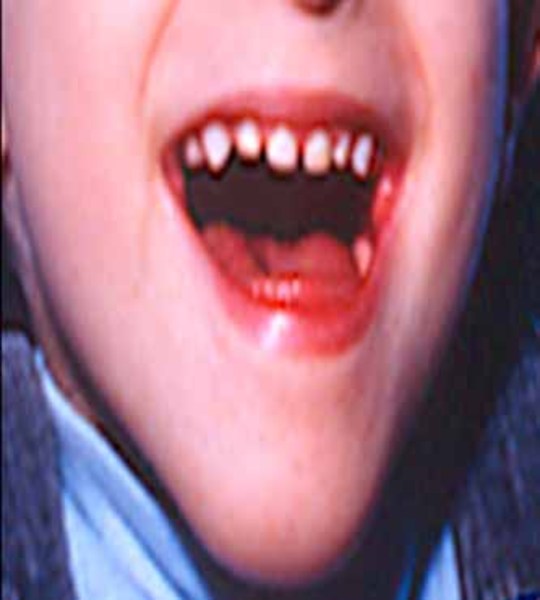
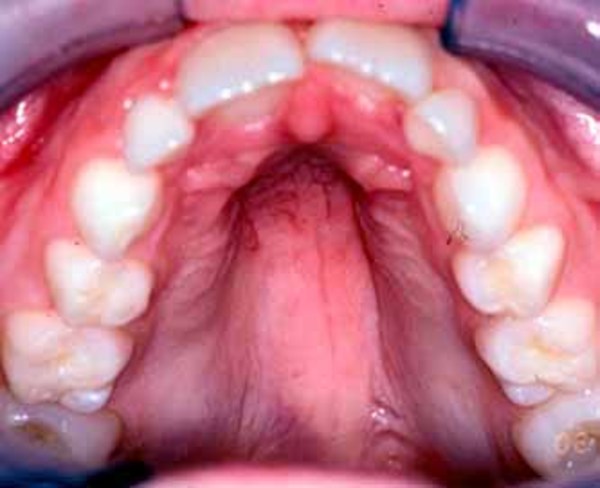
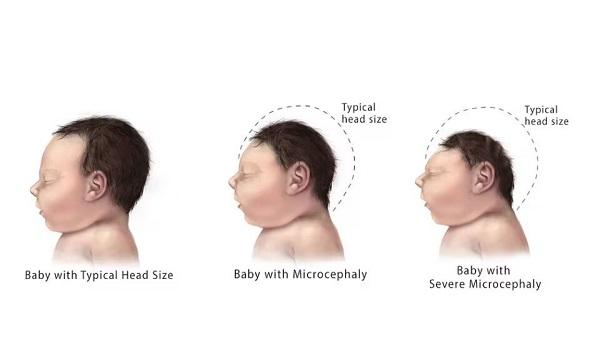
Other common features of CDKL5 deficiency disorder include repetitive hand movements (stereotypies), such as clapping, hand licking, and hand sucking; teeth grinding (bruxism); disrupted sleep; feeding difficulties; and gastrointestinal problems including constipation and backflow of acidic stomach contents into the esophagus (gastroesophageal reflux). Some affected individuals have episodes of irregular breathing. Distinctive facial features in some people with CDKL5 deficiency disorder include a high and broad forehead, large and deep-set eyes, a well-defined space between the nose and upper lip (philtrum), full lips, widely spaced teeth, and a high roof of the mouth (palate). Other physical differences can also occur, such as an unusually small head size (microcephaly), side-to-side curvature of the spine (scoliosis), and tapered fingers.


CDKL5 deficiency disorder was previously classified as an atypical form of Rett syndrome. These conditions have common features, including seizures, intellectual disability, and other problems with development. However, the signs and symptoms associated with CDKL5 deficiency disorder and its genetic cause are distinct from those of Rett syndrome, and CDKL5 deficiency disorder is now considered a separate condition.
Frequency
CDKL5 deficiency disorder appears to be a rare condition with an incidence of 1 in 40,000 to 60,000 newborns. About 90 percent of those diagnosed with CDKL5 deficiency disorder are girls.
Causes
As its name suggests, CDKL5 deficiency disorder is caused by variants (also known as mutations) in the CDKL5 gene. This gene provides instructions for making a protein that is essential for normal brain development and function.
Variants in the CDKL5 gene reduce the amount of functional CDKL5 protein or alter its activity in nerve cells (neurons). A shortage (deficiency) of CDKL5 or impairment of its function disrupts brain development, but it is unclear how these changes cause the specific features of CDKL5 deficiency disorder.

Inheritance
This condition is inherited in an X-linked dominant pattern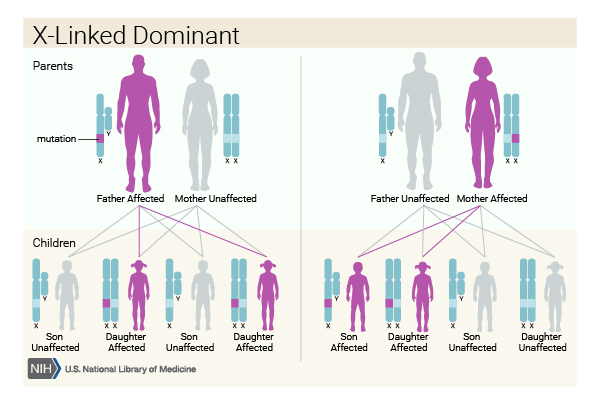 . The CDKL5 gene is located on the X chromosome, which is one of the two sex chromosomes
. The CDKL5 gene is located on the X chromosome, which is one of the two sex chromosomes . In females (who have two X chromosomes), a variant in one of the two copies of the CDKL5 gene in each cell causes the disorder. In males (who have only one X chromosome), a variant in the only copy of the gene causes the disorder. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. In females (who have two X chromosomes), a variant in one of the two copies of the CDKL5 gene in each cell causes the disorder. In males (who have only one X chromosome), a variant in the only copy of the gene causes the disorder. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Almost all cases of this condition result from new (de novo) variants in the CDKL5 gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Researchers suspect that the signs and symptoms of CDKL5 deficiency disorder vary in severity in part because of a process called X-inactivation. Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, such that each X chromosome is active in about half of the body cells. This means that about half of cells have an active X chromosome with a CDKL5 gene variant, and half have an active X chromosome without the variant. However, groups of cells that arise from a single original cell have the same copy of the X chromosome inactivated, so the distribution is not exactly half and half. The proportion of neurons in the brain that have the active X chromosome with the variant helps determine how severe the features of the condition are in a given individual. Females with a higher percentage of neurons with the variant have more severe signs and symptoms than females with a lower percentage of neurons with the variant.
Because males have only one X chromosome in each cell, the altered version of the CDKL5 gene is active in all cells. Affected males have no normal copies of the gene.
Other Names for This Condition
- CDKL5 deficiency
- CDKL5 disorder
- CDKL5 encephalopathy
- CDKL5-related epilepsy
- CDKL5-related epileptic encephalopathy
- Early infantile epileptic encephalopathy 2
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bahi-Buisson N, Bienvenu T. CDKL5-Related Disorders: From Clinical Description to Molecular Genetics. Mol Syndromol. 2012 Apr;2(3-5):137-152. doi: 10.1159/000331333. Epub 2011 Sep 13. Citation on PubMed or Free article on PubMed Central
- Bahi-Buisson N, Kaminska A, Boddaert N, Rio M, Afenjar A, Gerard M, Giuliano F, Motte J, Heron D, Morel MA, Plouin P, Richelme C, des Portes V, Dulac O, Philippe C, Chiron C, Nabbout R, Bienvenu T. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia. 2008 Jun;49(6):1027-37. doi: 10.1111/j.1528-1167.2007.01520.x. Epub 2008 Feb 7. Citation on PubMed
- Bahi-Buisson N, Villeneuve N, Caietta E, Jacquette A, Maurey H, Matthijs G, Van Esch H, Delahaye A, Moncla A, Milh M, Zufferey F, Diebold B, Bienvenu T. Recurrent mutations in the CDKL5 gene: genotype-phenotype relationships. Am J Med Genet A. 2012 Jul;158A(7):1612-9. doi: 10.1002/ajmg.a.35401. Epub 2012 Jun 7. Citation on PubMed
- Demarest ST, Olson HE, Moss A, Pestana-Knight E, Zhang X, Parikh S, Swanson LC, Riley KD, Bazin GA, Angione K, Niestroj LM, Lal D, Juarez-Colunga E, Benke TA. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia. 2019 Aug;60(8):1733-1742. doi: 10.1111/epi.16285. Epub 2019 Jul 16. Citation on PubMed
- Fehr S, Downs J, Ho G, de Klerk N, Forbes D, Christodoulou J, Williams S, Leonard H. Functional abilities in children and adults with the CDKL5 disorder. Am J Med Genet A. 2016 Nov;170(11):2860-2869. doi: 10.1002/ajmg.a.37851. Epub 2016 Aug 16. Citation on PubMed
- Fehr S, Wilson M, Downs J, Williams S, Murgia A, Sartori S, Vecchi M, Ho G, Polli R, Psoni S, Bao X, de Klerk N, Leonard H, Christodoulou J. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet. 2013 Mar;21(3):266-73. doi: 10.1038/ejhg.2012.156. Epub 2012 Aug 8. Citation on PubMed or Free article on PubMed Central
- Hector RD, Kalscheuer VM, Hennig F, Leonard H, Downs J, Clarke A, Benke TA, Armstrong J, Pineda M, Bailey MES, Cobb SR. CDKL5 variants: Improving our understanding of a rare neurologic disorder. Neurol Genet. 2017 Dec 15;3(6):e200. doi: 10.1212/NXG.0000000000000200. eCollection 2017 Dec. Citation on PubMed or Free article on PubMed Central
- Jahn J, Caliebe A, von Spiczak S, Boor R, Stefanova I, Stephani U, Helbig I, Muhle H. CDKL5 mutations as a cause of severe epilepsy in infancy: clinical and electroencephalographic long-term course in 4 patients. J Child Neurol. 2013 Jul;28(7):937-41. doi: 10.1177/0883073812451497. Epub 2012 Jul 25. Citation on PubMed
- Leonard H, Downs J, Benke TA, Swanson L, Olson H, Demarest S. CDKL5 deficiency disorder: clinical features, diagnosis, and management. Lancet Neurol. 2022 Jun;21(6):563-576. doi: 10.1016/S1474-4422(22)00035-7. Epub 2022 Apr 25. Citation on PubMed
- Mangatt M, Wong K, Anderson B, Epstein A, Hodgetts S, Leonard H, Downs J. Prevalence and onset of comorbidities in the CDKL5 disorder differ from Rett syndrome. Orphanet J Rare Dis. 2016 Apr 14;11:39. doi: 10.1186/s13023-016-0418-y. Citation on PubMed or Free article on PubMed Central
- Mori Y, Downs J, Wong K, Anderson B, Epstein A, Leonard H. Impacts of caring for a child with the CDKL5 disorder on parental wellbeing and family quality of life. Orphanet J Rare Dis. 2017 Jan 19;12(1):16. doi: 10.1186/s13023-016-0563-3. Citation on PubMed or Free article on PubMed Central
- Moseley BD, Dhamija R, Wirrell EC, Nickels KC. Historic, clinical, and prognostic features of epileptic encephalopathies caused by CDKL5 mutations. Pediatr Neurol. 2012 Feb;46(2):101-5. doi: 10.1016/j.pediatrneurol.2011.11.007. Citation on PubMed
- Muller A, Helbig I, Jansen C, Bast T, Guerrini R, Jahn J, Muhle H, Auvin S, Korenke GC, Philip S, Keimer R, Striano P, Wolf NI, Pust B, Thiels Ch, Fogarasi A, Waltz S, Kurlemann G, Kovacevic-Preradovic T, Ceulemans B, Schmitt B, Philippi H, Tarquinio D, Buerki S, von Stulpnagel C, Kluger G. Retrospective evaluation of low long-term efficacy of antiepileptic drugs and ketogenic diet in 39 patients with CDKL5-related epilepsy. Eur J Paediatr Neurol. 2016 Jan;20(1):147-51. doi: 10.1016/j.ejpn.2015.09.001. Epub 2015 Sep 10. Citation on PubMed
- Olson HE, Demarest ST, Pestana-Knight EM, Swanson LC, Iqbal S, Lal D, Leonard H, Cross JH, Devinsky O, Benke TA. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr Neurol. 2019 Aug;97:18-25. doi: 10.1016/j.pediatrneurol.2019.02.015. Epub 2019 Feb 23. Citation on PubMed
- Symonds JD, Zuberi SM, Stewart K, McLellan A, O'Regan M, MacLeod S, Jollands A, Joss S, Kirkpatrick M, Brunklaus A, Pilz DT, Shetty J, Dorris L, Abu-Arafeh I, Andrew J, Brink P, Callaghan M, Cruden J, Diver LA, Findlay C, Gardiner S, Grattan R, Lang B, MacDonnell J, McKnight J, Morrison CA, Nairn L, Slean MM, Stephen E, Webb A, Vincent A, Wilson M. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019 Aug 1;142(8):2303-2318. doi: 10.1093/brain/awz195. Citation on PubMed or Free article on PubMed Central
- Zhu YC, Xiong ZQ. Molecular and Synaptic Bases of CDKL5 Disorder. Dev Neurobiol. 2019 Jan;79(1):8-19. doi: 10.1002/dneu.22639. Epub 2018 Oct 19. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.