

Description
Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES) is a condition that mainly affects development of the eyelids. People with this condition have a narrowing of the eye opening (blepharophimosis), droopy eyelids (ptosis), and an upward fold of the skin of the lower eyelid near the inner corner of the eye (epicanthus inversus). In addition, there is an increased distance between the inner corners of the eyes (telecanthus). Because of these eyelid abnormalities, the eyelids cannot open fully, and vision may be limited.
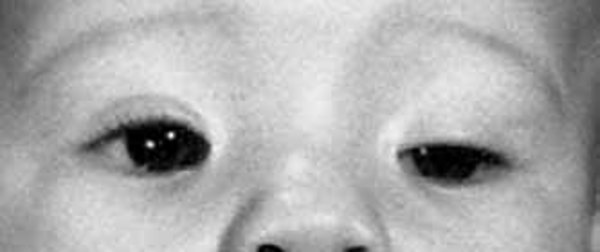
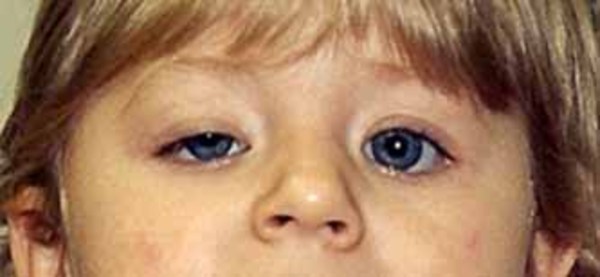
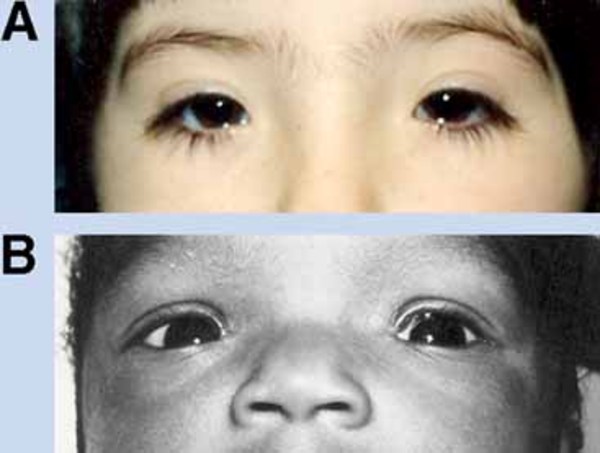
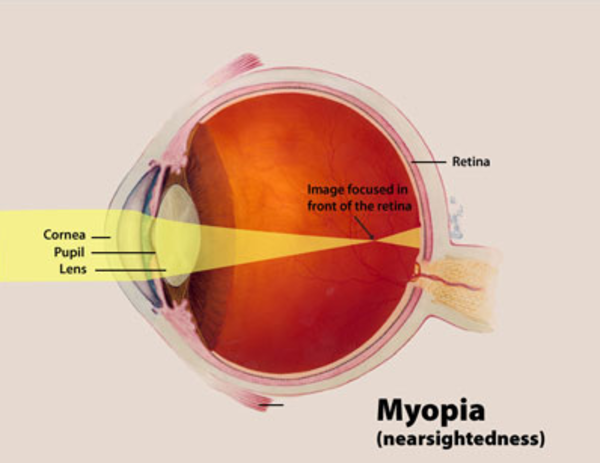
Other structures in the eyes and face may be mildly affected by BPES. Affected individuals are at an increased risk of developing vision problems such as nearsightedness (myopia) or farsightedness (hyperopia) beginning in childhood. They may also have eyes that do not point in the same direction (strabismus) or "lazy eye" (amblyopia) affecting one or both eyes. People with BPES may also have distinctive facial features including a broad nasal bridge, low-set ears, or a shortened distance between the nose and upper lip (a short philtrum).
There are two types of BPES, which are distinguished by their signs and symptoms. Both types I and II include the eyelid malformations and other facial features. Type I is also associated with an early loss of ovarian function (primary ovarian insufficiency) in women, which causes their menstrual periods to become less frequent and eventually stop before age 40. Primary ovarian insufficiency can lead to difficulty conceiving a child (subfertility) or a complete inability to conceive (infertility).
Frequency
The prevalence of BPES is unknown.
Causes
Mutations in the FOXL2 gene cause BPES types I and II. The FOXL2 gene provides instructions for making a protein that is active in the eyelids and ovaries. The FOXL2 protein is likely involved in the development of muscles in the eyelids. Before birth and in adulthood, the protein regulates the growth and development of certain ovarian cells and the breakdown of specific molecules.
It is difficult to predict the type of BPES that will result from the many FOXL2 gene mutations. However, mutations that result in a partial loss of FOXL2 protein function generally cause BPES type II. These mutations probably impair regulation of normal development of muscles in the eyelids, resulting in malformed eyelids that cannot open fully. Mutations that lead to a complete loss of FOXL2 protein function often cause BPES type I. These mutations impair the regulation of eyelid development as well as various activities in the ovaries, resulting in eyelid malformation and abnormally accelerated maturation of certain ovarian cells and the premature death of egg cells.
Inheritance
This condition is typically inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


Other Names for This Condition
- Blepharophimosis syndrome
- Blepharophimosis, ptosis, and epicanthus inversus
- BPES
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Allen CE, Rubin PA. Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES): clinical manifestation and treatment. Int Ophthalmol Clin. 2008 Spring;48(2):15-23. doi: 10.1097/IIO.0b013e3181694eee. No abstract available. Citation on PubMed
- Beysen D, De Jaegere S, Amor D, Bouchard P, Christin-Maitre S, Fellous M, Touraine P, Grix AW, Hennekam R, Meire F, Oyen N, Wilson LC, Barel D, Clayton-Smith J, de Ravel T, Decock C, Delbeke P, Ensenauer R, Ebinger F, Gillessen-Kaesbach G, Hendriks Y, Kimonis V, Laframboise R, Laissue P, Leppig K, Leroy BP, Miller DT, Mowat D, Neumann L, Plomp A, Van Regemorter N, Wieczorek D, Veitia RA, De Paepe A, De Baere E. Identification of 34 novel and 56 known FOXL2 mutations in patients with Blepharophimosis syndrome. Hum Mutat. 2008 Nov;29(11):E205-19. doi: 10.1002/humu.20819. Citation on PubMed
- Beysen D, De Paepe A, De Baere E. FOXL2 mutations and genomic rearrangements in BPES. Hum Mutat. 2009 Feb;30(2):158-69. doi: 10.1002/humu.20807. Citation on PubMed
- Beysen D, Raes J, Leroy BP, Lucassen A, Yates JR, Clayton-Smith J, Ilyina H, Brooks SS, Christin-Maitre S, Fellous M, Fryns JP, Kim JR, Lapunzina P, Lemyre E, Meire F, Messiaen LM, Oley C, Splitt M, Thomson J, Van de Peer Y, Veitia RA, De Paepe A, De Baere E. Deletions involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome. Am J Hum Genet. 2005 Aug;77(2):205-18. doi: 10.1086/432083. Epub 2005 Jun 16. Citation on PubMed or Free article on PubMed Central
- Choi KH, Kyung S, Oh SY. The factors influencing visual development in blepharophimosis-ptosis-epicanthus inversus syndrome. J Pediatr Ophthalmol Strabismus. 2006 Sep-Oct;43(5):285-8. doi: 10.3928/01913913-20060901-03. Citation on PubMed
- D'haene B, Nevado J, Pugeat M, Pierquin G, Lowry RB, Reardon W, Delicado A, Garcia-Minaur S, Palomares M, Courtens W, Stefanova M, Wallace S, Watkins W, Shelling AN, Wieczorek D, Veitia RA, De Paepe A, Lapunzina P, De Baere E. FOXL2 copy number changes in the molecular pathogenesis of BPES: unique cohort of 17 deletions. Hum Mutat. 2010 May;31(5):E1332-47. doi: 10.1002/humu.21233. Citation on PubMed
- Dipietromaria A, Benayoun BA, Todeschini AL, Rivals I, Bazin C, Veitia RA. Towards a functional classification of pathogenic FOXL2 mutations using transactivation reporter systems. Hum Mol Genet. 2009 Sep 1;18(17):3324-33. doi: 10.1093/hmg/ddp273. Epub 2009 Jun 10. Citation on PubMed
- Taylor A, Strike PW, Tyers AG. Blepharophimosis-ptosis-epicanthus inversus syndrome: objective analysis of surgical outcome in patients from a single unit. Clin Exp Ophthalmol. 2007 Apr;35(3):262-9. doi: 10.1111/j.1442-9071.2006.01448.x. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.