

Description
Barth syndrome is a rare condition characterized by an enlarged and weakened heart (dilated cardiomyopathy), weakness in muscles used for movement (skeletal myopathy), recurrent infections due to small numbers of white blood cells (neutropenia), and short stature. Barth syndrome occurs almost exclusively in males.
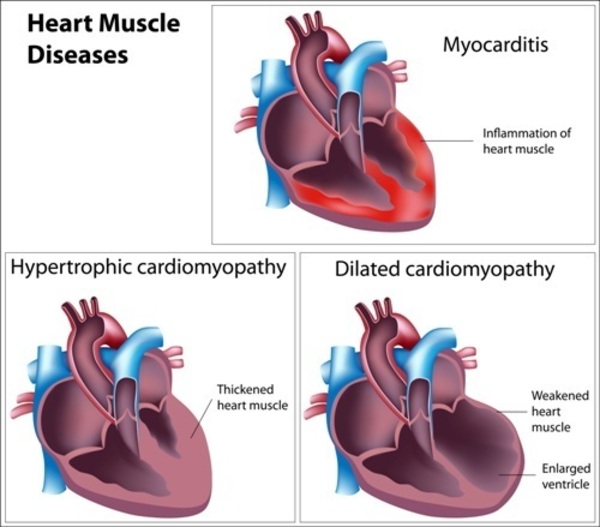
In males with Barth syndrome, dilated cardiomyopathy is often present at birth or develops within the first months of life. Over time, the heart muscle becomes increasingly weakened and is less able to pump blood. Individuals with Barth syndrome may have elastic fibers in place of muscle fibers in some areas of the heart muscle, which contributes to the cardiomyopathy. This condition is called endocardial fibroelastosis; it results in thickening of the muscle and impairs its ability to pump blood. In people with Barth syndrome, the heart problems can lead to heart failure. In rare cases, the cardiomyopathy gets better over time and affected individuals eventually have no symptoms of heart disease.
In Barth syndrome, skeletal myopathy, particularly of the muscles closest to the center of the body (proximal muscles), is usually noticeable from birth and causes low muscle tone (hypotonia). The muscle weakness often causes delay of motor skills such as crawling and walking. Additionally, affected individuals tend to experience extreme tiredness (fatigue) during strenuous physical activity.
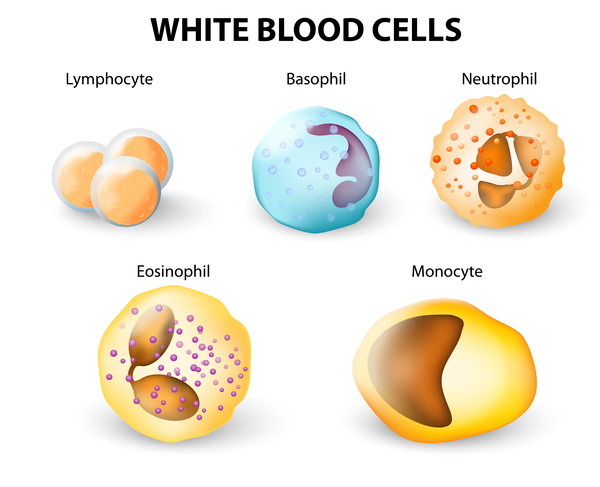
Most males with Barth syndrome have neutropenia. The levels of white blood cells can be consistently low (persistent), can vary from normal to low (intermittent), or can cycle between regular episodes of normal and low (cyclical). Neutropenia makes it more difficult for the body to fight off foreign invaders such as bacteria and viruses, so affected individuals have an increased risk of recurrent infections.
Newborns with Barth syndrome are often smaller than normal, and their growth continues to be slow throughout life. Some boys with this condition experience a growth spurt in puberty and are of average height as adults, but many men with Barth syndrome continue to have short stature in adulthood.
Males with Barth syndrome often have distinctive facial features including prominent cheeks. Affected individuals typically have normal intelligence but often have difficulty performing tasks involving math or visual-spatial skills such as puzzles.
Males with Barth syndrome have increased levels of a substance called 3-methylglutaconic acid in their blood and urine. The amount of the acid does not appear to influence the signs and symptoms of the condition. Barth syndrome is one of a group of metabolic disorders that can be diagnosed by the presence of increased levels of 3-methylglutaconic acid in urine (3-methylglutaconic aciduria).
Even though most features of Barth syndrome are present at birth or in infancy, affected individuals may not experience health problems until later in life. The age at which individuals with Barth syndrome display symptoms or are diagnosed varies greatly. The severity of signs and symptoms among affected individuals is also highly variable.
Males with Barth syndrome have a reduced life expectancy. Many affected children die of heart failure or infection in infancy or early childhood, but those who live into adulthood can survive into their late forties.
Frequency
Barth syndrome is estimated to affect 1 in 300,000 to 400,000 individuals worldwide. More than 150 cases have been described in the scientific literature.
Causes
Mutations in the TAFAZZIN gene cause Barth syndrome. The TAFAZZIN gene provides instructions for making a protein called tafazzin. This protein is located in structures called mitochondria, which are the energy-producing centers of cells. The tafazzin protein is involved in altering a fat (lipid) called cardiolipin, which plays critical roles in the mitochondrial inner membrane. Once altered by tafazzin, cardiolipin is key in maintaining mitochondrial shape, energy production, and protein transport within cells.
TAFAZZIN gene mutations result in the production of tafazzin proteins with little or no function. As a result, tafazzin cannot alter cardiolipin, and levels of functional cardiolipin are reduced. In addition, for unknown reasons, a variant of cardiolipin called monolysocardiolipin (MLCL) is formed. A lack of functional cardiolipin and an excess of MLCL are thought to impair normal mitochondrial shape and functions. Tissues with high energy demands, such as the heart and skeletal muscles, are most susceptible to cell death due to reduced energy production in mitochondria. Additionally, abnormally shaped mitochondria are found in affected white blood cells, which could affect their ability to grow (proliferate), mature (differentiate), and function, leading to neutropenia. Dysfunctional mitochondria likely lead to other signs and symptoms of Barth syndrome.
Inheritance
This condition is inherited in an X-linked recessive pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.


Other Names for This Condition
- 3 methylglutaconic aciduria, type II
- 3-methylglutaconic aciduria type 2
- BTHS
- Cardioskeletal myopathy with neutropenia and abnormal mitochondria
- DNAJC19 defect
- MGA type 2
- MGA type II
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Aprikyan AA, Khuchua Z. Advances in the understanding of Barth syndrome. Br J Haematol. 2013 May;161(3):330-8. doi: 10.1111/bjh.12271. Epub 2013 Feb 25. Citation on PubMed
- Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, Martin RP, Tsai-Goodman B, Garratt V, Ashworth M, Bowen VM, McCurdy KR, Damin MK, Spencer CT, Toth MJ, Kelley RI, Steward CG. Barth syndrome. Orphanet J Rare Dis. 2013 Feb 12;8:23. doi: 10.1186/1750-1172-8-23. Citation on PubMed or Free article on PubMed Central
- Ferreira C, Pierre G, Thompson R, Vernon H. Barth Syndrome. 2014 Oct 9 [updated 2020 Jul 9]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK247162/ Citation on PubMed
- Hastings R, Steward C, Tsai-Goodman B, Newbury-Ecob R. Dysmorphology of Barth syndrome. Clin Dysmorphol. 2009 Oct;18(4):185-7. doi: 10.1097/MCD.0b013e32832a9e62. Citation on PubMed
- Jefferies JL. Barth syndrome. Am J Med Genet C Semin Med Genet. 2013 Aug;163C(3):198-205. doi: 10.1002/ajmg.c.31372. Epub 2013 Jul 10. Citation on PubMed or Free article on PubMed Central
- Mazzocco MM, Henry AE, Kelly RI. Barth syndrome is associated with a cognitive phenotype. J Dev Behav Pediatr. 2007 Feb;28(1):22-30. doi: 10.1097/01.DBP.0000257519.79803.90. Citation on PubMed or Free article on PubMed Central
- Rigaud C, Lebre AS, Touraine R, Beaupain B, Ottolenghi C, Chabli A, Ansquer H, Ozsahin H, Di Filippo S, De Lonlay P, Borm B, Rivier F, Vaillant MC, Mathieu-Dramard M, Goldenberg A, Viot G, Charron P, Rio M, Bonnet D, Donadieu J. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J Rare Dis. 2013 May 8;8:70. doi: 10.1186/1750-1172-8-70. Citation on PubMed or Free article on PubMed Central
- Schlame M, Xu Y. The Function of Tafazzin, a Mitochondrial Phospholipid-Lysophospholipid Acyltransferase. J Mol Biol. 2020 Aug 21;432(18):5043-5051. doi: 10.1016/j.jmb.2020.03.026. Epub 2020 Mar 29. Citation on PubMed
- Steward CG, Groves SJ, Taylor CT, Maisenbacher MK, Versluys B, Newbury-Ecob RA, Ozsahin H, Damin MK, Bowen VM, McCurdy KR, Mackey MC, Bolyard AA, Dale DC. Neutropenia in Barth syndrome: characteristics, risks, and management. Curr Opin Hematol. 2019 Jan;26(1):6-15. doi: 10.1097/MOH.0000000000000472. Citation on PubMed
- Vernon HJ, Sandlers Y, McClellan R, Kelley RI. Clinical laboratory studies in Barth Syndrome. Mol Genet Metab. 2014 Jun;112(2):143-7. doi: 10.1016/j.ymgme.2014.03.007. Epub 2014 Mar 30. Citation on PubMed
- Wortmann SB, Duran M, Anikster Y, Barth PG, Sperl W, Zschocke J, Morava E, Wevers RA. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J Inherit Metab Dis. 2013 Nov;36(6):923-8. doi: 10.1007/s10545-012-9580-0. Epub 2013 Jan 8. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.