

Description
Ataxia with oculomotor apraxia is a condition characterized by problems with movement that worsen over time. The hallmark of this condition is poor coordination and balance (ataxia), which is often the first symptom. Most affected people also have oculomotor apraxia, which makes it difficult to move their eyes side-to-side. People with oculomotor apraxia have to turn their head to see things in their side (peripheral) vision.
There are several types of ataxia with oculomotor apraxia, the most common of which are types 1, 2, and 4. The types are very similar but are caused by mutations in different genes.
Type 1 begins around age 4. In addition to ataxia and oculomotor apraxia, affected individuals can have involuntary jerking movements (chorea) or muscle twitches (myoclonus); these movement problems tend to disappear over time. Individuals with this type may also develop muscle wasting in their hands and feet, which further impairs movement. As in all forms of ataxia with oculomotor apraxia, nearly all people with type 1 develop nerve abnormalities (neuropathy). Neuropathy impairs reflexes and leads to limb weakness and an inability to sense vibrations. Many individuals with ataxia with oculomotor apraxia require wheelchair assistance, typically 10 to 15 years after the start of movement problems.
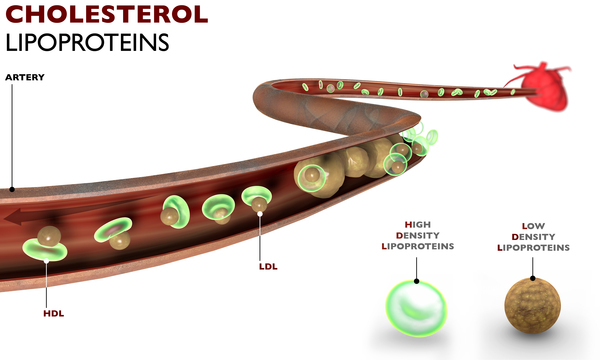
People with some types of ataxia with oculomotor apraxia may have characteristic blood abnormalities. Individuals with type 1 tend to have reduced amounts of a protein called albumin, which transports molecules in the blood. The shortage of albumin likely results in elevated levels of cholesterol circulating in the bloodstream. Increased cholesterol levels raise a person's risk of developing heart disease.
Ataxia with oculomotor apraxia type 2 usually begins around age 15. As in type 1, affected individuals may have chorea or myoclonus, although these movement problems persist throughout life in type 2. Neuropathy is also common in this type.
A key feature of ataxia with oculomotor apraxia type 2 is high amounts of a protein called alpha-fetoprotein (AFP) in the blood. (Raised levels of this protein are normally seen in the bloodstream of pregnant women.) Individuals with type 2 may also have high amounts of a protein called creatine phosphokinase (CPK) in their blood. This protein is normally found primarily in muscle tissue. The effect of abnormally high levels of AFP or CPK in people with ataxia with oculomotor apraxia type 2 is unknown. Although individuals with type 2 usually have normal albumin levels, cholesterol may be elevated.
Ataxia with oculomotor apraxia type 4 begins around age 4. In addition to ataxia and oculomotor apraxia, individuals with this type typically develop dystonia, which is involuntary, sustained muscle tensing that causes unusual positioning of body parts. Dystonia can be the first feature of the condition, and it tends to disappear gradually over time. Muscle wasting in the hands and feet and neuropathy are also common in individuals with type 4.
In ataxia with oculomotor apraxia type 4, albumin levels can be low, and cholesterol or AFP can be elevated. However, the amounts of these molecules are normal in many affected individuals.
Intelligence is usually not affected by ataxia with oculomotor apraxia, but some people with the condition have intellectual disability.
Frequency
Ataxia with oculomotor apraxia is a rare condition. Types 1 and 4 are most frequent in Portugal, and type 1 is also found in Japan. Type 2 is estimated to occur in 1 in 900,000 individuals worldwide. Type 3 has been found in only one family.
Causes
Mutations in the APTX, SETX, or PNKP gene cause ataxia with oculomotor apraxia types 1, 2, or 4, respectively. Mutations in another gene cause ataxia with oculomotor apraxia type 3.
The APTX, SETX, and PNKP genes provide instructions for making proteins that are involved in repairing damaged DNA. Mutations in any of these genes reduce the amount of functional protein produced from that gene. This shortage prevents the efficient repair of DNA damage, which leads to the accumulation of broken DNA strands. DNA breaks may be caused by potentially harmful molecules (called reactive oxygen species) produced during normal cellular functions, natural and medical radiation, or other environmental exposures. They may also occur when chromosomes exchange genetic material in preparation for cell division. DNA damage that is not repaired makes the cell unstable and can lead to cell death. It is thought that cell death has a particularly severe effect in the brain because the nervous system does not replace nerve cells that have been lost. The part of the brain involved in coordinating movements (the cerebellum) is especially at risk. It is thought that the loss of brain cells in the cerebellum causes the movement problems characteristic of ataxia with oculomotor apraxia.
Inheritance
All types of this condition are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Adult onset ataxia with oculomotor apraxia
- EAOH
- Early-onset ataxia with ocular motor apraxia and hypoalbuminemia
- SCAN2
- SCAR1
- Spinocerebellar ataxia with axonal neuropathy type 2
- Spinocerebellar ataxia, recessive, non-Friedreich type 1
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Ataxia - oculomotor apraxia type 4

- Genetic Testing Registry: Ataxia with oculomotor apraxia type 3
- Genetic Testing Registry: Ataxia, early-onset, with oculomotor apraxia and hypoalbuminemia
- Genetic Testing Registry: Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 2
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Al Tassan N, Khalil D, Shinwari J, Al Sharif L, Bavi P, Abduljaleel Z, Abu Dhaim N, Magrashi A, Bobis S, Ahmed H, Alahmed S, Bohlega S. A missense mutation in PIK3R5 gene in a family with ataxia and oculomotor apraxia. Hum Mutat. 2012 Feb;33(2):351-4. doi: 10.1002/humu.21650. Epub 2011 Dec 8. Citation on PubMed
- Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, Delaunoy JP, Fritsch M, Arning L, Synofzik M, Schols L, Sequeiros J, Goizet C, Marelli C, Le Ber I, Koht J, Gazulla J, De Bleecker J, Mukhtar M, Drouot N, Ali-Pacha L, Benhassine T, Chbicheb M, M'Zahem A, Hamri A, Chabrol B, Pouget J, Murphy R, Watanabe M, Coutinho P, Tazir M, Durr A, Brice A, Tranchant C, Koenig M. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009 Oct;132(Pt 10):2688-98. doi: 10.1093/brain/awp211. Epub 2009 Aug 20. Citation on PubMed
- Bras J, Alonso I, Barbot C, Costa MM, Darwent L, Orme T, Sequeiros J, Hardy J, Coutinho P, Guerreiro R. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet. 2015 Mar 5;96(3):474-9. doi: 10.1016/j.ajhg.2015.01.005. Epub 2015 Feb 26. Citation on PubMed or Free article on PubMed Central
- Choudry TN, Hilton-Jones D, Lennox G, Houlden H. Ataxia with oculomotor apraxia type 2: an evolving axonal neuropathy. Pract Neurol. 2018 Feb;18(1):52-56. doi: 10.1136/practneurol-2017-001711. Epub 2017 Dec 6. Citation on PubMed
- Criscuolo C, Chessa L, Di Giandomenico S, Mancini P, Sacca F, Grieco GS, Piane M, Barbieri F, De Michele G, Banfi S, Pierelli F, Rizzuto N, Santorelli FM, Gallosti L, Filla A, Casali C. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology. 2006 Apr 25;66(8):1207-10. doi: 10.1212/01.wnl.0000208402.10512.4a. Citation on PubMed
- Hoch NC, Hanzlikova H, Rulten SL, Tetreault M, Komulainen E, Ju L, Hornyak P, Zeng Z, Gittens W, Rey SA, Staras K, Mancini GM, McKinnon PJ, Wang ZQ, Wagner JD; Care4Rare Canada Consortium; Yoon G, Caldecott KW. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature. 2017 Jan 5;541(7635):87-91. doi: 10.1038/nature20790. Epub 2016 Dec 21. Citation on PubMed or Free article on PubMed Central
- Le Ber I, Brice A, Durr A. New autosomal recessive cerebellar ataxias with oculomotor apraxia. Curr Neurol Neurosci Rep. 2005 Sep;5(5):411-7. doi: 10.1007/s11910-005-0066-4. Citation on PubMed
- Le Ber I, Moreira MC, Rivaud-Pechoux S, Chamayou C, Ochsner F, Kuntzer T, Tardieu M, Said G, Habert MO, Demarquay G, Tannier C, Beis JM, Brice A, Koenig M, Durr A. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain. 2003 Dec;126(Pt 12):2761-72. doi: 10.1093/brain/awg283. Epub 2003 Sep 23. Citation on PubMed
- O'Connor E, Vandrovcova J, Bugiardini E, Chelban V, Manole A, Davagnanam I, Wiethoff S, Pittman A, Lynch DS, Efthymiou S, Marino S, Manzur AY, Roberts M, Hanna MG, Houlden H, Matthews E, Wood NW. Mutations in XRCC1 cause cerebellar ataxia and peripheral neuropathy. J Neurol Neurosurg Psychiatry. 2018 Nov;89(11):1230-1232. doi: 10.1136/jnnp-2017-317581. Epub 2018 Feb 22. No abstract available. Citation on PubMed
- Renaud M, Moreira MC, Ben Monga B, Rodriguez D, Debs R, Charles P, Chaouch M, Ferrat F, Laurencin C, Vercueil L, Mallaret M, M'Zahem A, Pacha LA, Tazir M, Tilikete C, Ollagnon E, Ochsner F, Kuntzer T, Jung HH, Beis JM, Netter JC, Djamshidian A, Bower M, Bottani A, Walsh R, Murphy S, Reiley T, Bieth E, Roelens F, Poll-The BT, Lourenco CM, Jardim LB, Straussberg R, Landrieu P, Roze E, Thobois S, Pouget J, Guissart C, Goizet C, Durr A, Tranchant C, Koenig M, Anheim M. Clinical, Biomarker, and Molecular Delineations and Genotype-Phenotype Correlations of Ataxia With Oculomotor Apraxia Type 1. JAMA Neurol. 2018 Apr 1;75(4):495-502. doi: 10.1001/jamaneurol.2017.4373. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.