

Description
Amyotrophic lateral sclerosis (ALS) is a progressive disease that affects motor neurons, which are specialized nerve cells that control muscle movement. These nerve cells are found in the spinal cord and the brain. In ALS, motor neurons die (atrophy) over time, leading to muscle weakness, a loss of muscle mass, and an inability to control movement.
There are many different types of ALS; these types are distinguished by their signs and symptoms and their genetic cause or lack of clear genetic association. Most people with ALS have a form of the condition that is described as sporadic, which means it occurs in people with no apparent history of the disorder in their family. People with sporadic ALS usually first develop features of the condition in their late fifties or early sixties. A small proportion of people with ALS, estimated at 5 to 10 percent, have a family history of ALS or a related condition called frontotemporal dementia (FTD), which is a progressive brain disorder that affects personality, behavior, and language. The signs and symptoms of familial ALS typically first appear in one's late forties or early fifties. Rarely, people with familial ALS develop symptoms in childhood or their teenage years. These individuals have a rare form of the disorder known as juvenile ALS.
The first signs and symptoms of ALS may be so subtle that they are overlooked. The earliest symptoms include muscle twitching, cramping, stiffness, or weakness. Affected individuals may develop slurred speech (dysarthria) and, later, difficulty chewing or swallowing (dysphagia). Many people with ALS experience malnutrition because of reduced food intake due to dysphagia and an increase in their body's energy demands (metabolism) due to prolonged illness. Muscles become weaker as the disease progresses, and arms and legs begin to look thinner as muscle tissue atrophies. Individuals with ALS eventually lose muscle strength and the ability to walk. Affected individuals eventually become wheelchair-dependent and increasingly require help with personal care and other activities of daily living. Over time, muscle weakness causes affected individuals to lose the use of their hands and arms. Breathing becomes difficult because the muscles of the respiratory system weaken. Most people with ALS die from respiratory failure within 2 to 10 years after the signs and symptoms of ALS first appear; however, disease progression varies widely among affected individuals.
Approximately 20 percent of individuals with ALS also develop FTD. Changes in personality and behavior may make it difficult for affected individuals to interact with others in a socially appropriate manner. Communication skills worsen as the disease progresses. It is unclear how the development of ALS and FTD are related. Individuals who develop both conditions are diagnosed as having ALS-FTD.
A rare form of ALS that often runs in families is known as ALS-parkinsonism-dementia complex (ALS-PDC). This disorder is characterized by the signs and symptoms of ALS, in addition to a pattern of movement abnormalities known as parkinsonism, and a progressive loss of intellectual function (dementia). Signs of parkinsonism include unusually slow movements (bradykinesia), stiffness, and tremors. Affected members of the same family can have different combinations of signs and symptoms.
Frequency
About 5,000 people in the United States are diagnosed with ALS each year. Worldwide, this disorder occurs in 2 to 5 per 100,000 individuals. Only a small percentage of cases have a known genetic cause.
Among the Chamorro people of Guam and people from the Kii Peninsula of Japan, ALS-PDC can be 100 times more frequent than ALS is in other populations. ALS-PDC has not been reported outside of these populations.
Causes
Mutations in several genes can cause familial ALS and contribute to the development of sporadic ALS. Mutations in the C9orf72 gene account for 30 to 40 percent of familial ALS in the United States and Europe. Worldwide, SOD1 gene mutations cause 15 to 20 percent of familial ALS, and TARDBP and FUS gene mutations each account for about 5 percent of cases. The other genes that have been associated with familial ALS each account for a small proportion of cases. It is estimated that 60 percent of individuals with familial ALS have an identified genetic mutation. The cause of the condition in the remaining individuals is unknown.
The C9orf72, SOD1, TARDBP, and FUS genes are key to the normal functioning of motor neurons and other cells. It is unclear how mutations in these genes contribute to the death of motor neurons, but it is thought that motor neurons are more sensitive to disruptions in function because of their large size. Most motor neurons affected by ALS have a buildup of protein clumps (aggregates); however, it is unknown whether these aggregates are involved in causing ALS or are a byproduct of the dying cell.
In some cases of familial ALS due to mutations in other genes, studies have identified the mechanisms that lead to ALS. Some gene mutations lead to a disruption in the development of axons, the specialized extensions of nerve cells (such as motor neurons) that transmit nerve impulses. The altered axons may impair transmission of impulses from nerves to muscles, leading to muscle weakness and atrophy. Other mutations lead to a slowing in the transport of materials needed for the proper function of axons in motor neurons, eventually causing the motor neurons to die. Additional gene mutations prevent the breakdown of toxic substances, leading to their buildup in nerve cells. The accumulation of toxic substances can damage motor neurons and eventually cause cell death. In some cases of ALS, it is unknown how the gene mutation causes the condition.

The cause of sporadic ALS is largely unknown but probably involves a combination of genetic and environmental factors. Variations in many genes, including the previously mentioned genes involved in transmission of nerve impulses and transportation of materials within neurons, increase the risk of developing ALS. Gene mutations that are risk factors for ALS may add, delete, or change DNA building blocks (nucleotides), resulting in the production of a protein with an altered or reduced function. While genetic variations have been associated with sporadic ALS, not all genetic factors have been identified and it is unclear how most genetic changes influence the development of the disease. People with a gene variation that increases their risk of ALS likely require additional genetic and environmental triggers to develop the disorder.
Inheritance
About 90 to 95 percent of ALS cases are sporadic, which means they are not inherited.
An estimated 5 to 10 percent of ALS is familial and caused by mutations in one of several genes. The pattern of inheritance varies depending on the gene involved. Most cases are inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition. Some people who inherit a familial genetic mutation known to cause ALS never develop features of the condition. (This situation is known as reduced penetrance.) It is unclear why some people with a mutated gene develop the disease and other people with a mutated gene do not.

Less frequently, ALS is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Because an affected person's parents are not affected, autosomal recessive ALS is often mistaken for sporadic ALS even though it is caused by a familial genetic mutation.

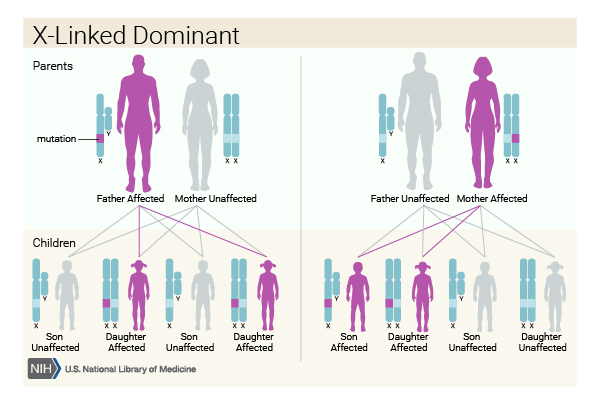
Very rarely, ALS is inherited in an X-linked dominant pattern. X-linked conditions occur when the gene associated with the condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. In most cases, males tend to develop the disease earlier and have a decreased life expectancy compared with females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Other Names for This Condition
- ALS
- Amyotrophic lateral sclerosis with dementia
- Charcot disease
- Dementia with amyotrophic lateral sclerosis
- Lou Gehrig disease
- Motor neuron disease, amyotrophic lateral sclerosis
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- AMYOTROPHIC LATERAL SCLEROSIS 1; ALS1
- AMYOTROPHIC LATERAL SCLEROSIS-PARKINSONISM/DEMENTIA COMPLEX 1
- FRONTOTEMPORAL DEMENTIA AND/OR AMYOTROPHIC LATERAL SCLEROSIS 1; FTDALS1
- AMYOTROPHIC LATERAL SCLEROSIS 5, JUVENILE; ALS5
- AMYOTROPHIC LATERAL SCLEROSIS 2, JUVENILE; ALS2
- FRONTOTEMPORAL DEMENTIA AND/OR AMYOTROPHIC LATERAL SCLEROSIS 7; FTDALS7
- AMYOTROPHIC LATERAL SCLEROSIS 15 WITH OR WITHOUT FRONTOTEMPORAL DEMENTIA; ALS15
- AMYOTROPHIC LATERAL SCLEROSIS 4, JUVENILE; ALS4
- AMYOTROPHIC LATERAL SCLEROSIS 8; ALS8
- AMYOTROPHIC LATERAL SCLEROSIS 6 WITH OR WITHOUT FRONTOTEMPORAL DEMENTIA; ALS6
- AMYOTROPHIC LATERAL SCLEROSIS 7; ALS7
- AMYOTROPHIC LATERAL SCLEROSIS 3; ALS3
- AMYOTROPHIC LATERAL SCLEROSIS 9; ALS9
- AMYOTROPHIC LATERAL SCLEROSIS 10 WITH OR WITHOUT FRONTOTEMPORAL DEMENTIA; ALS10
- AMYOTROPHIC LATERAL SCLEROSIS 11; ALS11
- AMYOTROPHIC LATERAL SCLEROSIS 16, JUVENILE; ALS16
- AMYOTROPHIC LATERAL SCLEROSIS 12 WITH OR WITHOUT FRONTOTEMPORAL DEMENTIA; ALS12
- AMYOTROPHIC LATERAL SCLEROSIS 19; ALS19
- FRONTOTEMPORAL DEMENTIA AND/OR AMYOTROPHIC LATERAL SCLEROSIS 2; FTDALS2
- FRONTOTEMPORAL DEMENTIA AND/OR AMYOTROPHIC LATERAL SCLEROSIS 6; FTDALS6
- AMYOTROPHIC LATERAL SCLEROSIS 20; ALS20
- AMYOTROPHIC LATERAL SCLEROSIS 22 WITH OR WITHOUT FRONTOTEMPORAL DEMENTIA; ALS22
- FRONTOTEMPORAL DEMENTIA AND/OR AMYOTROPHIC LATERAL SCLEROSIS 3; FTDALS3
- FRONTOTEMPORAL DEMENTIA AND/OR AMYOTROPHIC LATERAL SCLEROSIS 4; FTDALS4
- AMYOTROPHIC LATERAL SCLEROSIS 18; ALS18
Scientific Articles on PubMed
References
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011 Oct 11;7(11):603-15. doi: 10.1038/nrneurol.2011.150. Citation on PubMed
- Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006 Jan;6(1):37-46. doi: 10.1007/s11910-996-0008-9. Citation on PubMed
- Chio A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, Sendtner M, Brunetti M, Ossola I, Calvo A, Pugliatti M, Sotgiu MA, Murru MR, Marrosu MG, Marrosu F, Marinou K, Mandrioli J, Sola P, Caponnetto C, Mancardi G, Mandich P, La Bella V, Spataro R, Conte A, Monsurro MR, Tedeschi G, Pisano F, Bartolomei I, Salvi F, Lauria Pinter G, Simone I, Logroscino G, Gambardella A, Quattrone A, Lunetta C, Volanti P, Zollino M, Penco S, Battistini S; ITALSGEN consortium; Renton AE, Majounie E, Abramzon Y, Conforti FL, Giannini F, Corbo M, Sabatelli M. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain. 2012 Mar;135(Pt 3):784-93. doi: 10.1093/brain/awr366. Citation on PubMed or Free article on PubMed Central
- Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014 Jul 1;23(13):3579-95. doi: 10.1093/hmg/ddu068. Epub 2014 Feb 18. Erratum In: Hum Mol Genet. 2017 Oct 15;26(20):4093-4094. Citation on PubMed or Free article on PubMed Central
- Fecto F, Siddique T. Making connections: pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementia. J Mol Neurosci. 2011 Nov;45(3):663-75. doi: 10.1007/s12031-011-9637-9. Epub 2011 Sep 7. Citation on PubMed
- Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011 Nov;7(11):616-30. doi: 10.1038/nrneurol.2011.152. Citation on PubMed
- Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011 Mar 12;377(9769):942-55. doi: 10.1016/S0140-6736(10)61156-7. Epub 2011 Feb 4. Citation on PubMed
- Lattante S, Ciura S, Rouleau GA, Kabashi E. Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD). Trends Genet. 2015 May;31(5):263-73. doi: 10.1016/j.tig.2015.03.005. Epub 2015 Apr 10. Citation on PubMed
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013 Aug 7;79(3):416-38. doi: 10.1016/j.neuron.2013.07.033. Citation on PubMed or Free article on PubMed Central
- Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2015 May 14;1607:75-93. doi: 10.1016/j.brainres.2014.10.009. Epub 2014 Oct 12. Citation on PubMed
- McGeer PL, Steele JC. The ALS/PDC syndrome of Guam: potential biomarkers for an enigmatic disorder. Prog Neurobiol. 2011 Dec;95(4):663-9. doi: 10.1016/j.pneurobio.2011.04.001. Epub 2011 Apr 20. Citation on PubMed
- National Institute of Neurological Disorders and Stroke
- Paganoni S, Macklin EA, Lee A, Murphy A, Chang J, Zipf A, Cudkowicz M, Atassi N. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener. 2014 Sep;15(5-6):453-6. doi: 10.3109/21678421.2014.903974. Epub 2014 Jul 1. Citation on PubMed or Free article on PubMed Central
- Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014 Jan;17(1):17-23. doi: 10.1038/nn.3584. Epub 2013 Dec 26. Citation on PubMed or Free article on PubMed Central
- Siddique N, Siddique T. Amyotrophic Lateral Sclerosis Overview. 2001 Mar 23 [updated 2023 Sep 28]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1450/ Citation on PubMed
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009 Feb 27;323(5918):1208-1211. doi: 10.1126/science.1165942. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.