

Description
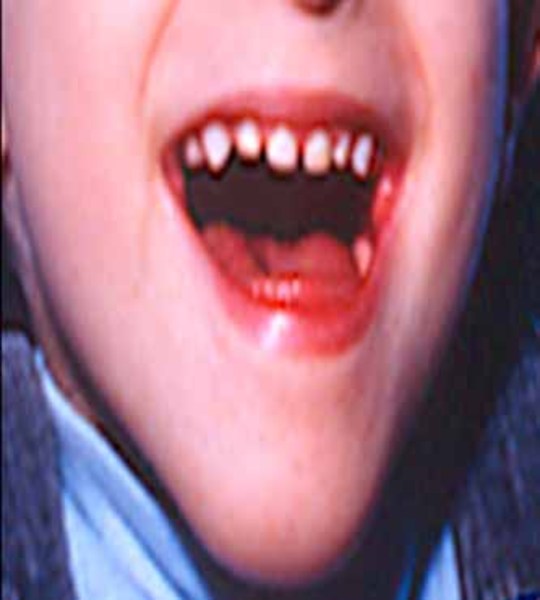
Alpha-mannosidosis is a rare inherited disorder that causes problems in many organs and tissues of the body. Affected individuals may have intellectual disability, distinctive facial features, and skeletal abnormalities. Characteristic facial features can include a large head, prominent forehead, low hairline, rounded eyebrows, large ears, flattened bridge of the nose, protruding jaw, widely spaced teeth, overgrown gums, and large tongue. The skeletal abnormalities that can occur in this disorder include reduced bone density (osteopenia), thickening of the bones at the top of the skull (calvaria), deformations of the bones in the spine (vertebrae), knock knees, and deterioration of the bones and joints.
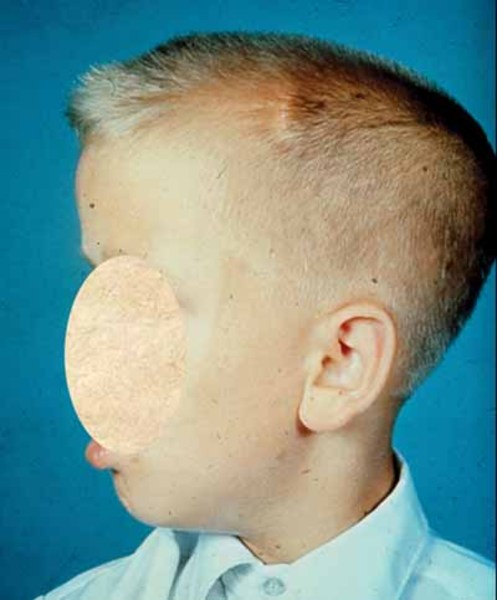
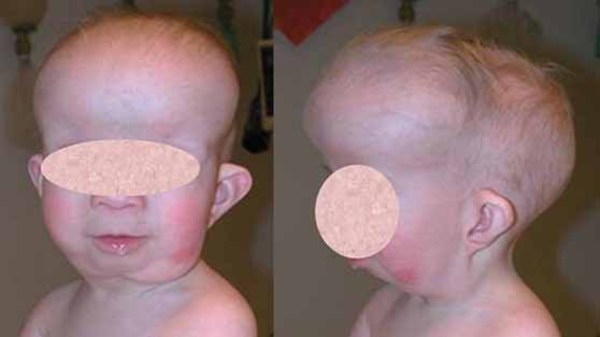
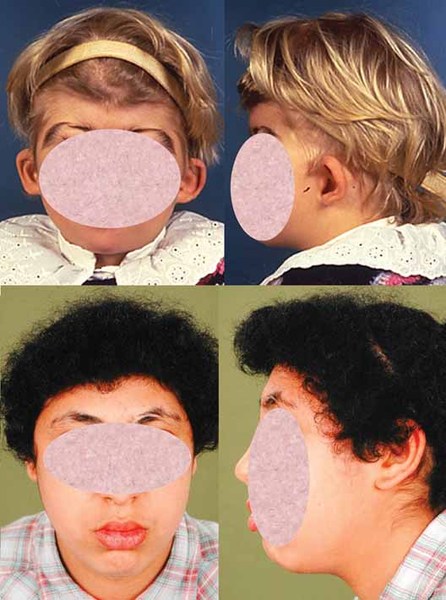


Affected individuals may also experience difficulty in coordinating movements (ataxia); muscle weakness (myopathy); delay in developing motor skills such as sitting and walking; speech impairments; increased risk of infections; enlargement of the liver and spleen (hepatosplenomegaly); a buildup of fluid in the brain (hydrocephalus); hearing loss; and a clouding of the lens of the eye (cataract). Some people with alpha-mannosidosis experience psychiatric symptoms such as depression, anxiety, or hallucinations; episodes of psychiatric disturbance may be triggered by stressors such as having undergone surgery, emotional upset, or changes in routine.
The signs and symptoms of alpha-mannosidosis can range from mild to severe. The disorder may appear in infancy with rapid progression and severe neurological deterioration. Individuals with this early-onset form of alpha-mannosidosis often do not survive past childhood. In the most severe cases, an affected fetus may die before birth. Other individuals with alpha-mannosidosis experience milder signs and symptoms that appear later and progress more slowly. People with later-onset alpha-mannosidosis may survive into their fifties. The mildest cases may be detected only through laboratory testing and result in few if any symptoms.
Frequency
Alpha-mannosidosis is estimated to occur in approximately 1 in 500,000 people worldwide.
Causes
Mutations in the MAN2B1 gene cause alpha-mannosidosis. This gene provides instructions for making the enzyme alpha-mannosidase. This enzyme works in the lysosomes, which are compartments that digest and recycle materials in the cell. Within lysosomes, the enzyme helps break down complexes of sugar molecules (oligosaccharides) attached to certain proteins (glycoproteins). In particular, alpha-mannosidase helps break down oligosaccharides containing a sugar molecule called mannose.
Mutations in the MAN2B1 gene interfere with the ability of the alpha-mannosidase enzyme to perform its role in breaking down mannose-containing oligosaccharides. These oligosaccharides accumulate in the lysosomes and cause cells to malfunction and eventually die. Tissues and organs are damaged by the abnormal accumulation of oligosaccharides and the resulting cell death, leading to the characteristic features of alpha-mannosidosis.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Alpha-D-mannosidosis
- Alpha-mannosidase B deficiency
- Alpha-mannosidase deficiency
- Deficiency of alpha-mannosidase
- Lysosomal alpha B mannosidosis
- Lysosomal alpha-D-mannosidase deficiency
- Mannosidosis
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Beck M, Olsen KJ, Wraith JE, Zeman J, Michalski JC, Saftig P, Fogh J, Malm D. Natural history of alpha mannosidosis a longitudinal study. Orphanet J Rare Dis. 2013 Jun 20;8:88. doi: 10.1186/1750-1172-8-88. Citation on PubMed or Free article on PubMed Central
- Grewal SS, Shapiro EG, Krivit W, Charnas L, Lockman LA, Delaney KA, Davies SM, Wenger DA, Rimell FL, Abel S, Grovas AC, Orchard PJ, Wagner JE, Peters C. Effective treatment of alpha-mannosidosis by allogeneic hematopoietic stem cell transplantation. J Pediatr. 2004 May;144(5):569-73. doi: 10.1016/j.jpeds.2004.01.025. Citation on PubMed
- Gutschalk A, Harting I, Cantz M, Springer C, Rohrschneider K, Meinck HM. Adult alpha-mannosidosis: clinical progression in the absence of demyelination. Neurology. 2004 Nov 9;63(9):1744-6. doi: 10.1212/01.wnl.0000143057.25471.4f. Citation on PubMed
- Hansen G, Berg T, Riise Stensland HM, Heikinheimo P, Klenow H, Evjen G, Nilssen O, Tollersrud OK. Intracellular transport of human lysosomal alpha-mannosidase and alpha-mannosidosis-related mutants. Biochem J. 2004 Jul 15;381(Pt 2):537-46. doi: 10.1042/BJ20031499. Citation on PubMed or Free article on PubMed Central
- Lyons MJ, Wood T, Espinoza L, Stensland HM, Holden KR. Early onset alpha-mannosidosis with slow progression in three Hispanic males. Dev Med Child Neurol. 2007 Nov;49(11):854-7. doi: 10.1111/j.1469-8749.2007.00854.x. Erratum In: Dev Med Child Neurol. 2008 Jan;50(1):32. Citation on PubMed
- Malm D, Nilssen O. Alpha-Mannosidosis. 2001 Oct 11 [updated 2019 Jul 18]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1396/ Citation on PubMed
- Malm D, Nilssen O. Alpha-mannosidosis. Orphanet J Rare Dis. 2008 Jul 23;3:21. doi: 10.1186/1750-1172-3-21. Citation on PubMed or Free article on PubMed Central
- Malm D, Pantel J, Linaker OM. Psychiatric symptoms in alpha-mannosidosis. J Intellect Disabil Res. 2005 Nov;49(Pt 11):865-71. doi: 10.1111/j.1365-2788.2005.00765.x. Citation on PubMed
- Pittis MG, Montalvo AL, Heikinheimo P, Sbaragli M, Balducci C, Persichetti E, Van Maldergem L, Filocamo M, Bembi B, Beccari T. Funtional characterization of four novel MAN2B1 mutations causing juvenile onset alpha-mannosidosis. Clin Chim Acta. 2007 Jan;375(1-2):136-9. doi: 10.1016/j.cca.2006.06.034. Epub 2006 Jul 6. Citation on PubMed
- Sbaragli M, Bibi L, Pittis MG, Balducci C, Heikinheimo P, Ricci R, Antuzzi D, Parini R, Spaccini L, Bembi B, Beccari T. Identification and characterization of five novel MAN2B1 mutations in Italian patients with alpha-mannosidosis. Hum Mutat. 2005 Mar;25(3):320. doi: 10.1002/humu.9310. Citation on PubMed
- Sun H, Wolfe JH. Recent progress in lysosomal alpha-mannosidase and its deficiency. Exp Mol Med. 2001 Mar 31;33(1):1-7. doi: 10.1038/emm.2001.1. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.