

Description
Alpers-Huttenlocher syndrome is one of the most severe of a group of conditions called the POLG-related disorders. The conditions in this group feature a range of similar signs and symptoms involving muscle-, nerve-, and brain-related functions. Alpers-Huttenlocher syndrome typically becomes apparent in children between ages 2 and 4. People with this condition usually have three characteristic features: recurrent seizures that do not improve with treatment (intractable epilepsy), loss of mental and movement abilities (psychomotor regression), and liver disease.
People with Alpers-Huttenlocher syndrome usually have additional signs and symptoms. Most have problems with coordination and balance (ataxia) and disturbances in nerve function (neuropathy). Neuropathy can lead to abnormal or absent reflexes (areflexia). In addition, affected individuals may develop weak muscle tone (hypotonia) that worsens until they lose the ability to control their muscles and movement. Some people with Alpers-Huttenlocher syndrome lose the ability to walk, sit, or feed themselves. Other movement-related symptoms in affected individuals can include involuntary muscle twitches (myoclonus), uncontrollable movements of the limbs (choreoathetosis), or a pattern of movement abnormalities known as parkinsonism.
Affected individuals may have other brain-related signs and symptoms. Migraine headaches, often with visual sensations or auras, are common. Additionally, people with this condition may have decreased brain function that is demonstrated as sleepiness, inability to concentrate, irritability, or loss of language skills or memory. Some people with the condition may lose their eyesight or hearing. People with Alpers-Huttenlocher syndrome can survive from a few months to more than 10 years after the condition first appears.
Frequency
The prevalence of Alpers-Huttenlocher syndrome is approximately 1 in 100,000 individuals.
Causes
Alpers-Huttenlocher syndrome is caused by mutations in the POLG gene. This gene provides instructions for making one part, the alpha subunit, of a protein called polymerase gamma (pol γ). Pol γ functions in mitochondria, which are structures within cells that use oxygen to convert the energy from food into a form cells can use. Mitochondria each contain a small amount of DNA, known as mitochondrial DNA (mtDNA), which is essential for the normal function of these structures. Pol γ "reads" sequences of mtDNA and uses them as templates to produce new copies of mtDNA in a process called DNA replication.
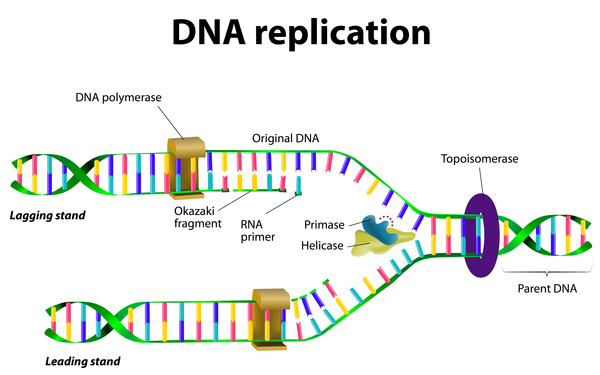
Most POLG gene mutations change single protein building blocks (amino acids) in the alpha subunit of pol γ. These changes result in a mutated pol γ that has a reduced ability to replicate DNA. Although the mechanism is unknown, mutations in the POLG gene often result in a reduced number of copies of mtDNA (mtDNA depletion), particularly in muscle, brain, and liver cells. MtDNA depletion causes a decrease in cellular energy, which could account for the signs and symptoms of Alpers-Huttenlocher syndrome.

A mutation in the POLG gene has not been identified in approximately 13 percent of people diagnosed with Alpers-Huttenlocher syndrome. Researchers are working to identify other genes that may be responsible for the condition.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Alpers diffuse degeneration of cerebral gray matter with hepatic cirrhosis
- Alpers disease
- Alpers progressive infantile poliodystrophy
- Alpers syndrome
- Diffuse cerebral sclerosis of Schilder
- Progressive sclerosing poliodystrophy
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Chan SS, Longley MJ, Copeland WC. The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J Biol Chem. 2005 Sep 9;280(36):31341-6. doi: 10.1074/jbc.M506762200. Epub 2005 Jul 16. Citation on PubMed
- Cohen BH, Chinnery PF, Copeland WC. POLG-Related Disorders. 2010 Mar 16 [updated 2024 Feb 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK26471/ Citation on PubMed
- Milone M, Massie R. Polymerase gamma 1 mutations: clinical correlations. Neurologist. 2010 Mar;16(2):84-91. doi: 10.1097/NRL.0b013e3181c78a89. Citation on PubMed
- Moraes CT, Shanske S, Tritschler HJ, Aprille JR, Andreetta F, Bonilla E, Schon EA, DiMauro S. mtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet. 1991 Mar;48(3):492-501. Citation on PubMed or Free article on PubMed Central
- Nguyen KV, Sharief FS, Chan SS, Copeland WC, Naviaux RK. Molecular diagnosis of Alpers syndrome. J Hepatol. 2006 Jul;45(1):108-16. doi: 10.1016/j.jhep.2005.12.026. Epub 2006 Feb 20. Citation on PubMed
- Rocher C, Taanman JW, Pierron D, Faustin B, Benard G, Rossignol R, Malgat M, Pedespan L, Letellier T. Influence of mitochondrial DNA level on cellular energy metabolism: implications for mitochondrial diseases. J Bioenerg Biomembr. 2008 Apr;40(2):59-67. doi: 10.1007/s10863-008-9130-5. Epub 2008 Apr 16. Citation on PubMed
- Stumpf JD, Copeland WC. Mitochondrial DNA replication and disease: insights from DNA polymerase gamma mutations. Cell Mol Life Sci. 2011 Jan;68(2):219-33. doi: 10.1007/s00018-010-0530-4. Epub 2010 Oct 8. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.