

Description
Alagille syndrome is a genetic disorder that can affect the liver, heart, and other parts of the body.
One of the major features of Alagille syndrome is liver damage caused by abnormalities in the bile ducts. These ducts carry bile (which helps to digest fats) from the liver to the gallbladder and small intestine. In Alagille syndrome, the bile ducts may be narrow, malformed, and reduced in number (bile duct paucity). As a result, bile builds up in the liver and causes scarring that prevents the liver from working properly to eliminate wastes from the bloodstream. Signs and symptoms arising from liver damage in Alagille syndrome may include a yellowish tinge in the skin and the whites of the eyes (jaundice), itchy skin, and deposits of cholesterol in the skin (xanthomas).
Alagille syndrome is also associated with several heart problems, including impaired blood flow from the heart into the lungs (pulmonic stenosis). Pulmonic stenosis may occur along with a hole between the two lower chambers of the heart (ventricular septal defect) and other heart abnormalities. This combination of heart defects is called tetralogy of Fallot.
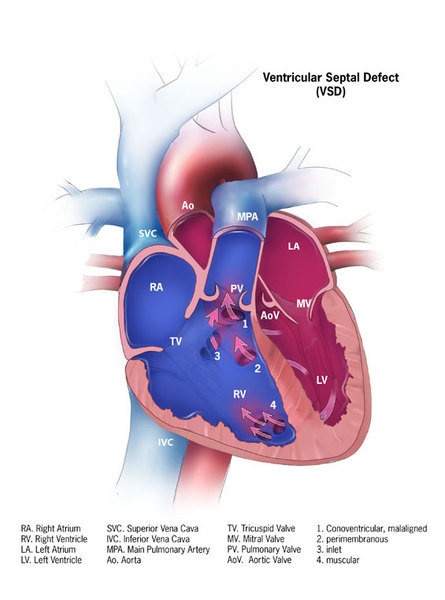
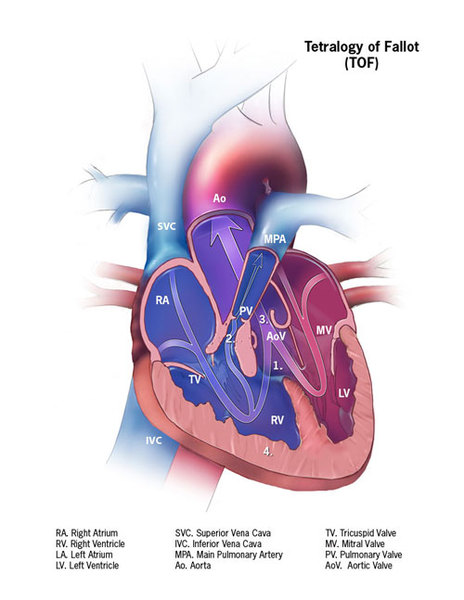
People with Alagille syndrome may have distinctive facial features including a broad, prominent forehead; deep-set eyes; and a small, pointed chin. The disorder may also affect the blood vessels within the brain and spinal cord (central nervous system) and the kidneys. Affected individuals may have an unusual butterfly shape of the bones of the spinal column (vertebrae) that can be seen in an x-ray.
Problems associated with Alagille syndrome generally become evident in infancy or early childhood. The severity of the disorder varies among affected individuals, even within the same family. Symptoms range from so mild as to go unnoticed to severe heart and/or liver disease requiring transplantation.
Some people with Alagille syndrome may have isolated signs of the disorder, such as a heart defect like tetralogy of Fallot, or a characteristic facial appearance. These individuals do not have liver disease or other features typical of the disorder.
Frequency
The estimated prevalence of Alagille syndrome is 1 in 70,000 newborns. This figure is based on diagnoses of liver disease in infants, and may be an underestimation because some people with Alagille syndrome do not develop liver disease during infancy.
Causes
In more than 90 percent of cases, mutations in the JAG1 gene cause Alagille syndrome. Another 7 percent of individuals with Alagille syndrome have small deletions of genetic material on chromosome 20 that include the JAG1 gene. A few people with Alagille syndrome have mutations in a different gene, called NOTCH2. The JAG1 and NOTCH2 genes provide instructions for making proteins that fit together to trigger interactions called Notch signaling between neighboring cells during embryonic development. This signaling influences how the cells are used to build body structures in the developing embryo. Changes in either the JAG1 gene or NOTCH2 gene probably disrupt the Notch signaling pathway. As a result, errors may occur during development, especially affecting the bile ducts, heart, spinal column, and certain facial features.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered or deleted gene in each cell is sufficient to cause the disorder.
In approximately 30 to 50 percent of cases, an affected person inherits the mutation or deletion from one affected parent. Other cases result from new mutations in the gene or new deletions of genetic material on chromosome 20 that occur as random events during the formation of reproductive cells (eggs or sperm) or in early fetal development. These cases occur in people with no history of the disorder in their family.


Other Names for This Condition
- Alagille's syndrome
- Alagille-Watson syndrome
- Arteriohepatic dysplasia (AHD)
- Cardiovertebral syndrome
- Cholestasis with peripheral pulmonary stenosis
- Hepatic ductular hypoplasia
- Hepatofacioneurocardiovertebral syndrome
- Paucity of interlobular bile ducts
- Watson-Miller syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Berniczei-Royko A, Chalas R, Mitura I, Nagy K, Prussak E. Medical and dental management of Alagille syndrome: a review. Med Sci Monit. 2014 Mar 24;20:476-80. doi: 10.12659/MSM.890577. Citation on PubMed or Free article on PubMed Central
- Boyer-Di Ponio J, Wright-Crosnier C, Groyer-Picard MT, Driancourt C, Beau I, Hadchouel M, Meunier-Rotival M. Biological function of mutant forms of JAGGED1 proteins in Alagille syndrome: inhibitory effect on Notch signaling. Hum Mol Genet. 2007 Nov 15;16(22):2683-92. doi: 10.1093/hmg/ddm222. Epub 2007 Aug 24. Citation on PubMed
- Hartley JL, Gissen P, Kelly DA. Alagille syndrome and other hereditary causes of cholestasis. Clin Liver Dis. 2013 May;17(2):279-300. doi: 10.1016/j.cld.2012.12.004. Citation on PubMed
- Kamath BM, Spinner NB, Rosenblum ND. Renal involvement and the role of Notch signalling in Alagille syndrome. Nat Rev Nephrol. 2013 Jul;9(7):409-18. doi: 10.1038/nrneph.2013.102. Epub 2013 Jun 11. Citation on PubMed
- Kim BJ, Fulton AB. The genetics and ocular findings of Alagille syndrome. Semin Ophthalmol. 2007 Oct-Dec;22(4):205-10. doi: 10.1080/08820530701745108. Citation on PubMed
- McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006 Jul;79(1):169-73. doi: 10.1086/505332. Epub 2006 May 10. Citation on PubMed or Free article on PubMed Central
- Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in alagille syndrome. Hum Mutat. 2001;17(1):18-33. doi: 10.1002/1098-1004(2001)17:13.0.CO;2-T. Citation on PubMed
- Spinner NB, Loomes KM, Krantz ID, Gilbert MA. Alagille Syndrome. 2000 May 19 [updated 2024 Jan 4]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1273/ Citation on PubMed
- Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012 Mar;20(3):251-7. doi: 10.1038/ejhg.2011.181. Epub 2011 Sep 21. Citation on PubMed or Free article on PubMed Central
- Warthen DM, Moore EC, Kamath BM, Morrissette JJ, Sanchez-Lara PA, Piccoli DA, Krantz ID, Spinner NB. Jagged1 (JAG1) mutations in Alagille syndrome: increasing the mutation detection rate. Hum Mutat. 2006 May;27(5):436-43. doi: 10.1002/humu.20310. Erratum In: Hum Mutat. 2013 Feb;34(2):408. Sanchez, P [corrected to Sanchez-Lara, P A]. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.