

Description
21-hydroxylase deficiency is an inherited disorder that affects the adrenal glands. The adrenal glands are located on top of the kidneys and produce a variety of hormones that regulate many essential functions in the body. In people with 21-hydroxylase deficiency, the adrenal glands produce excess androgens, which are male sex hormones.
There are three types of 21-hydroxylase deficiency. Two types are classic forms, known as the salt-wasting and simple virilizing types. The third type is called the non-classic type. The salt-wasting type is the most severe, the simple virilizing type is less severe, and the non-classic type is the least severe form.
Males and females with either classic form of 21-hydroxylase deficiency tend to have an early growth spurt, but their final adult height is usually shorter than others in their family. Additionally, affected individuals may have a reduced ability to have biological children (decreased fertility). Females may also develop excessive body hair growth (hirsutism), male pattern baldness, and irregular menstruation.
Approximately 75 percent of individuals with classic 21-hydroxylase deficiency have the salt-wasting type. Hormone production is extremely low in this form of the disorder. Affected individuals lose large amounts of sodium in their urine, which can be life-threatening in early infancy. Babies with the salt-wasting type can experience poor feeding, weight loss, dehydration, and vomiting. Individuals with the simple virilizing form do not experience salt loss.
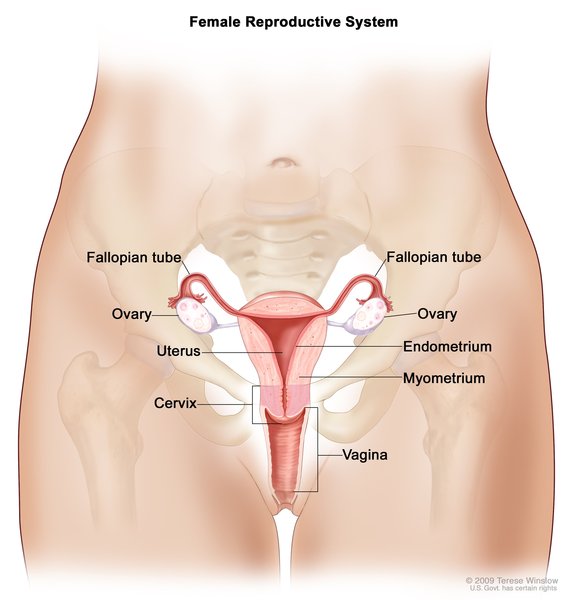
In both the salt-wasting and simple virilizing forms of this disorder, females typically have external genitalia that do not look clearly male or female. Males usually have male-typical genitalia but the testes may be small.
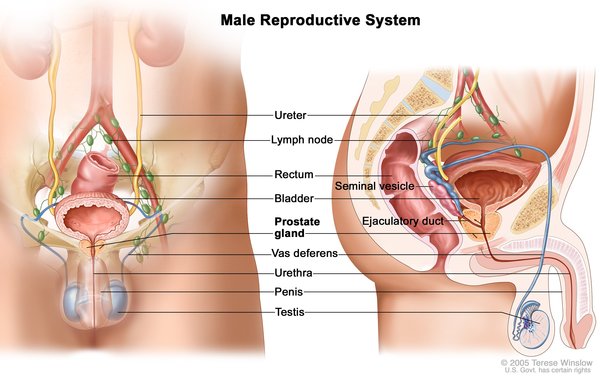
Females with the non-classic type of 21-hydroxylase deficiency have female-typical genitalia. As affected females get older, they may experience hirsutism, male pattern baldness, irregular menstruation, and decreased fertility. Males with the non-classic type may have early beard growth and small testes. Some individuals with this type of 21-hydroxylase deficiency have no symptoms of the disorder.
Frequency
The classic forms of 21-hydroxylase deficiency occur in 1 in 15,000 newborns. The prevalence of the non-classic form of 21-hydroxylase deficiency is estimated to be 1 in 1,000 individuals. The prevalence of both classic and non-classic forms varies among different ethnic populations.
21-hydroxylase deficiency is one of a group of disorders known as congenital adrenal hyperplasias that impair hormone production and disrupt sexual development. 21-hydroxylase deficiency is responsible for about 95 percent of all cases of congenital adrenal hyperplasia.
Causes
Mutations in the CYP21A2 gene cause 21-hydroxylase deficiency. The CYP21A2 gene provides instructions for making an enzyme called 21-hydroxylase. This enzyme is found in the adrenal glands, where it plays a role in producing hormones called cortisol and aldosterone. Cortisol has numerous functions, such as maintaining blood sugar (glucose) levels, protecting the body from stress, and suppressing inflammation. Aldosterone is sometimes called the salt-retaining hormone because it regulates the amount of salt retained by the kidneys. The retention of salt affects fluid levels in the body and blood pressure.
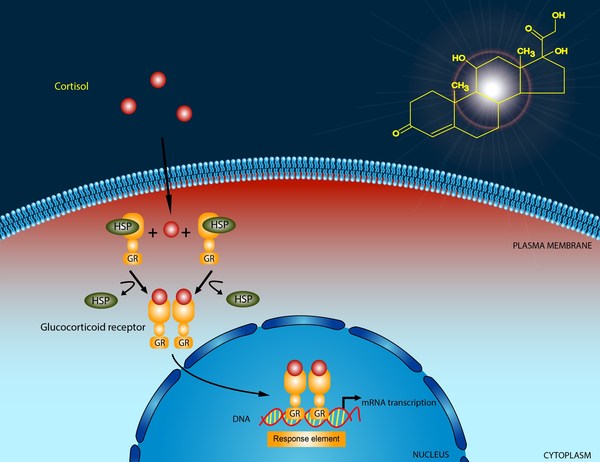

21-hydroxylase deficiency is caused by a shortage (deficiency) of the 21-hydroxylase enzyme. When 21-hydroxylase is lacking, substances that are usually used to form cortisol and aldosterone instead build up in the adrenal glands and are converted to androgens. The excess production of androgens leads to abnormalities of sexual development in people with 21-hydroxylase deficiency. A lack of aldosterone production contributes to the salt loss in people with the salt-wasting form of this condition.
The amount of functional 21-hydroxylase enzyme determines the severity of the disorder. Individuals with the salt-wasting type have CYP21A2 mutations that result in a completely nonfunctional enzyme. People with the simple virilizing type of this condition have CYP21A2 gene mutations that allow the production of low levels of functional enzyme. Individuals with the non-classic type of this disorder have CYP21A2 mutations that result in the production of reduced amounts of the enzyme, but more enzyme than either of the other types.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- CAH1
- Congenital adrenal hyperplasia 1
- Congenital adrenal hyperplasia due to 21 hydroxylase deficiency
- CYP21 deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bidet M, Bellanne-Chantelot C, Galand-Portier MB, Tardy V, Billaud L, Laborde K, Coussieu C, Morel Y, Vaury C, Golmard JL, Claustre A, Mornet E, Chakhtoura Z, Mowszowicz I, Bachelot A, Touraine P, Kuttenn F. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. J Clin Endocrinol Metab. 2009 May;94(5):1570-8. doi: 10.1210/jc.2008-1582. Epub 2009 Feb 10. Citation on PubMed
- Gidlof S, Falhammar H, Thilen A, von Dobeln U, Ritzen M, Wedell A, Nordenstrom A. One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study. Lancet Diabetes Endocrinol. 2013 Sep;1(1):35-42. doi: 10.1016/S2213-8587(13)70007-X. Epub 2013 Feb 26. Erratum In: Lancet Diabetes Endocrinol. 2013 Aug;1 Suppl 1:s22. Citation on PubMed
- Huynh T, McGown I, Cowley D, Nyunt O, Leong GM, Harris M, Cotterill AM. The clinical and biochemical spectrum of congenital adrenal hyperplasia secondary to 21-hydroxylase deficiency. Clin Biochem Rev. 2009 May;30(2):75-86. Citation on PubMed or Free article on PubMed Central
- Joint LWPES/ESPE CAH Working Group.. Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J Clin Endocrinol Metab. 2002 Sep;87(9):4048-53. doi: 10.1210/jc.2002-020611. No abstract available. Citation on PubMed
- Kruse B, Riepe FG, Krone N, Bosinski HA, Kloehn S, Partsch CJ, Sippell WG, Monig H. Congenital adrenal hyperplasia - how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. 2004 Jul;112(7):343-55. doi: 10.1055/s-2004-821013. Citation on PubMed
- Marumudi E, Khadgawat R, Surana V, Shabir I, Joseph A, Ammini AC. Diagnosis and management of classical congenital adrenal hyperplasia. Steroids. 2013 Aug;78(8):741-6. doi: 10.1016/j.steroids.2013.04.007. Epub 2013 Apr 25. Citation on PubMed
- Nimkarn S, Lin-Su K, New MI. Steroid 21 hydroxylase deficiency congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. 2009 Dec;38(4):699-718. doi: 10.1016/j.ecl.2009.08.001. Citation on PubMed
- Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. 2013 Aug;78(8):747-50. doi: 10.1016/j.steroids.2013.04.010. Epub 2013 Apr 28. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.