

Description
2-hydroxyglutaric aciduria is a condition that causes progressive damage to the brain. The major types of this disorder are called D-2-hydroxyglutaric aciduria (D-2-HGA), L-2-hydroxyglutaric aciduria (L-2-HGA), and combined D,L-2-hydroxyglutaric aciduria (D,L-2-HGA).
The main features of D-2-HGA are delayed development, seizures, weak muscle tone (hypotonia), and abnormalities in the largest part of the brain (the cerebrum), which controls many important functions such as muscle movement, speech, vision, thinking, emotion, and memory. Researchers have described two subtypes of D-2-HGA, type I and type II. The two subtypes are distinguished by their genetic cause and pattern of inheritance, although they also have some differences in signs and symptoms. Type II tends to begin earlier and often causes more severe health problems than type I. Type II may also be associated with a weakened and enlarged heart (cardiomyopathy), a feature that is typically not found with type I.
L-2-HGA particularly affects a region of the brain called the cerebellum, which is involved in coordinating movements. As a result, many affected individuals have problems with balance and muscle coordination (ataxia). Additional features of L-2-HGA can include delayed development, seizures, speech difficulties, and an unusually large head (macrocephaly). Typically, signs and symptoms of this disorder begin during infancy or early childhood. The disorder worsens over time, usually leading to severe disability by early adulthood.
Combined D,L-2-HGA causes severe brain abnormalities that become apparent in early infancy. Affected infants have severe seizures, weak muscle tone (hypotonia), and breathing and feeding problems. They usually survive only into infancy or early childhood.
Frequency
2-hydroxyglutaric aciduria is a rare disorder. D-2-HGA and L-2-HGA have each been reported to affect fewer than 150 individuals worldwide. Combined D,L-2-HGA appears to be even rarer, with only about a dozen reported cases.
Causes
The different types of 2-hydroxyglutaric aciduria result from mutations in several genes. D-2-HGA type I is caused by mutations in the D2HGDH gene; type II is caused by mutations in the IDH2 gene. L-2-HGA results from mutations in the L2HGDH gene. Combined D,L-2-HGA is caused by mutations in the SLC25A1 gene.
The D2HGDH and L2HGDH genes provide instructions for making enzymes that are found in mitochondria, which are the energy-producing centers within cells. The enzymes break down compounds called D-2-hydroxyglutarate and L-2-hydroxyglutarate, respectively, as part of a series of reactions that produce energy for cell activities. Mutations in either of these genes lead to a shortage of functional enzyme, which allows D-2-hydroxyglutarate or L-2-hydroxyglutarate to build up in cells. At high levels, these compounds can damage cells and lead to cell death. Brain cells appear to be the most vulnerable to the toxic effects of these compounds, which may explain why the signs and symptoms of D-2-HGA type I and L-2-HGA primarily involve the brain.
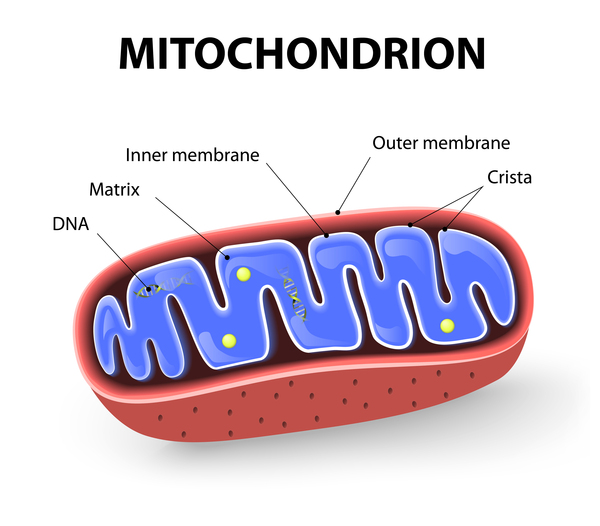
The IDH2 gene provides instructions for making an enzyme in mitochondria that normally produces a different compound. When the enzyme is altered by mutations, it takes on a new, abnormal function: production of the potentially toxic compound D-2-hydroxyglutarate. The resulting excess of this compound damages brain cells, leading to the signs and symptoms of D-2-HGA type II. It is unclear why an accumulation of D-2-hydroxyglutarate may be associated with cardiomyopathy in some people with this form of the condition.
The SLC25A1 gene provides instructions for making a protein that transports certain molecules, such as citrate, in and out of mitochondria. Mutations in the SLC25A1 gene reduce the protein's function, which prevents it from carrying out this transport. Through processes that are not fully understood, a loss of this transport allows both D-2-hydroxyglutarate and L-2-hydroxyglutarate to build up, which damages brain cells. Researchers suspect that an imbalance of other molecules, particularly citrate, also contributes to the severe signs and symptoms of combined D,L-2-HGA.
Inheritance
D-2-HGA type I, L-2-HGA, and combined D,L-2-HGA all have an autosomal recessive pattern of inheritance, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

D-2-HGA type II is considered an autosomal dominant disorder because one copy of the altered gene in each cell is sufficient to cause the condition. The disorder typically results from a new mutation in the IDH2 gene and occurs in people with no history of the condition in their family.

Other Names for This Condition
- 2-HGA
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Kranendijk M, Struys EA, Gibson KM, Wickenhagen WV, Abdenur JE, Buechner J, Christensen E, de Kremer RD, Errami A, Gissen P, Gradowska W, Hobson E, Islam L, Korman SH, Kurczynski T, Maranda B, Meli C, Rizzo C, Sansaricq C, Trefz FK, Webster R, Jakobs C, Salomons GS. Evidence for genetic heterogeneity in D-2-hydroxyglutaric aciduria. Hum Mutat. 2010 Mar;31(3):279-83. doi: 10.1002/humu.21186. Citation on PubMed
- Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012 Jul;35(4):571-87. doi: 10.1007/s10545-012-9462-5. Epub 2012 Mar 6. Citation on PubMed or Free article on PubMed Central
- Kranendijk M, Struys EA, van Schaftingen E, Gibson KM, Kanhai WA, van der Knaap MS, Amiel J, Buist NR, Das AM, de Klerk JB, Feigenbaum AS, Grange DK, Hofstede FC, Holme E, Kirk EP, Korman SH, Morava E, Morris A, Smeitink J, Sukhai RN, Vallance H, Jakobs C, Salomons GS. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010 Oct 15;330(6002):336. doi: 10.1126/science.1192632. Epub 2010 Sep 16. Citation on PubMed
- Muntau AC, Roschinger W, Merkenschlager A, van der Knaap MS, Jakobs C, Duran M, Hoffmann GF, Roscher AA. Combined D-2- and L-2-hydroxyglutaric aciduria with neonatal onset encephalopathy: a third biochemical variant of 2-hydroxyglutaric aciduria? Neuropediatrics. 2000 Jun;31(3):137-40. doi: 10.1055/s-2000-7497. Citation on PubMed
- Nota B, Struys EA, Pop A, Jansen EE, Fernandez Ojeda MR, Kanhai WA, Kranendijk M, van Dooren SJ, Bevova MR, Sistermans EA, Nieuwint AW, Barth M, Ben-Omran T, Hoffmann GF, de Lonlay P, McDonald MT, Meberg A, Muntau AC, Nuoffer JM, Parini R, Read MH, Renneberg A, Santer R, Strahleck T, van Schaftingen E, van der Knaap MS, Jakobs C, Salomons GS. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am J Hum Genet. 2013 Apr 4;92(4):627-31. doi: 10.1016/j.ajhg.2013.03.009. Citation on PubMed or Free article on PubMed Central
- Rzem R, Veiga-da-Cunha M, Noel G, Goffette S, Nassogne MC, Tabarki B, Scholler C, Marquardt T, Vikkula M, Van Schaftingen E. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc Natl Acad Sci U S A. 2004 Nov 30;101(48):16849-54. doi: 10.1073/pnas.0404840101. Epub 2004 Nov 17. Citation on PubMed or Free article on PubMed Central
- Struys EA, Salomons GS, Achouri Y, Van Schaftingen E, Grosso S, Craigen WJ, Verhoeven NM, Jakobs C. Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria. Am J Hum Genet. 2005 Feb;76(2):358-60. doi: 10.1086/427890. Epub 2004 Dec 17. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.