

Description
17q12 deletion syndrome is a condition that results from the deletion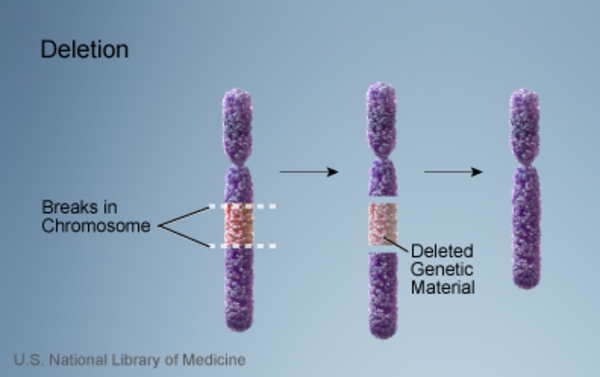 of a small piece of chromosome 17 in each cell. The deletion occurs on the long (q) arm of the chromosome at a position designated q12.
of a small piece of chromosome 17 in each cell. The deletion occurs on the long (q) arm of the chromosome at a position designated q12.
The signs and symptoms of 17q12 deletion syndrome vary widely, even among affected members of the same family. Among the more common features associated with this chromosomal change are problems with development or function of the kidneys and urinary system . These abnormalities range from very severe malformations, leading to kidney failure before birth, to mild or no problems with kidney and urinary tract function. Fluid-filled sacs (cysts) in the kidneys are particularly common. Many affected individuals also develop a form of diabetes called maturity-onset diabetes of the young type 5 (MODY5), which is caused by a malfunction of certain cells in the pancreas
. These abnormalities range from very severe malformations, leading to kidney failure before birth, to mild or no problems with kidney and urinary tract function. Fluid-filled sacs (cysts) in the kidneys are particularly common. Many affected individuals also develop a form of diabetes called maturity-onset diabetes of the young type 5 (MODY5), which is caused by a malfunction of certain cells in the pancreas . MODY5 usually appears in adolescence or early adulthood, most often before age 25. The combination of kidney cysts and MODY5 is sometimes referred to as renal cysts and diabetes (RCAD) syndrome.
. MODY5 usually appears in adolescence or early adulthood, most often before age 25. The combination of kidney cysts and MODY5 is sometimes referred to as renal cysts and diabetes (RCAD) syndrome.
About half of people with 17q12 deletion syndrome have delayed development (particularly speech and language delays), intellectual disability, or behavioral or psychiatric disorders. Neurodevelopmental and psychiatric conditions that have been reported in people with 17q12 deletion syndrome include autism spectrum disorder (which affects social interaction and communication), schizophrenia, anxiety, and bipolar disorder.
Less commonly, 17q12 deletion syndrome also causes abnormalities of the eyes, liver, brain, genitalia, and other body systems. Some females with this chromosomal change have Mayer-Rokitansky-Küster-Hauser syndrome, which is characterized by underdevelopment or absence of the vagina and uterus . 17q12 deletion syndrome is also sometimes associated with subtle differences in facial features.
. 17q12 deletion syndrome is also sometimes associated with subtle differences in facial features.
Frequency
The worldwide prevalence of 17q12 deletion syndrome is unknown, although the condition appears to be rare. One study estimated that 17q12 deletion syndrome occurs in 1 in 14,500 people in Iceland.
Causes
Most people with 17q12 deletion syndrome are missing about 1.4 million DNA building blocks (base pairs), also written as 1.4 megabases (Mb), at position q12 on chromosome 17. This deletion affects one of the two copies of chromosome 17 in each cell.
The deleted segment is surrounded by short, repeated sequences of DNA that make the segment prone to rearrangement during cell division. The rearrangement can lead to missing or extra copies of DNA at 17q12. (The presence of an extra copy of this segment is called a 17q12 duplication.)
The chromosome segment most commonly deleted in people with 17q12 deletion syndrome contains 15 genes. The loss of two genes in particular, HNF1B and LHX1, is thought to underlie some of the features of 17q12 deletion syndrome. Studies suggest that a loss of one copy of the HNF1B gene in each cell causes the kidney and urinary tract abnormalities, as well as abnormalities of the pancreas that underlie diabetes. The loss of one copy of LHX1 is thought to contribute to intellectual disability, behavioral and psychiatric conditions, and Mayer-Rokitansky-Küster-Hauser syndrome. The loss of other genes in the deleted region may also influence the signs and symptoms that can occur in 17q12 deletion syndrome.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the chromosomal deletion in each cell is sufficient to cause the disorder.
Most cases of 17q12 deletion syndrome result from a new (de novo) chromosomal deletion and occur in people with no history of the disorder in their family. Less commonly, an affected person inherits the deletion from one affected parent.


Other Names for This Condition
- 17q12 chromosomal microdeletion
- 17q12 microdeletion
- 17q12 recurrent deletion syndrome
- Deletion 17q12
- Recurrent genomic rearrangement in chromosome 17q12
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Laffargue F, Bourthoumieu S, Llanas B, Baudouin V, Lahoche A, Morin D, Bessenay L, De Parscau L, Cloarec S, Delrue MA, Taupiac E, Dizier E, Laroche C, Bahans C, Yardin C, Lacombe D, Guigonis V. Towards a new point of view on the phenotype of patients with a 17q12 microdeletion syndrome. Arch Dis Child. 2015 Mar;100(3):259-64. doi: 10.1136/archdischild-2014-306810. Epub 2014 Oct 16. Citation on PubMed
- Mefford HC, Clauin S, Sharp AJ, Moller RS, Ullmann R, Kapur R, Pinkel D, Cooper GM, Ventura M, Ropers HH, Tommerup N, Eichler EE, Bellanne-Chantelot C. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet. 2007 Nov;81(5):1057-69. doi: 10.1086/522591. Epub 2007 Sep 26. Citation on PubMed or Free article on PubMed Central
- Mitchel MW, Moreno-De-Luca D, Myers SM, Levy RV, Turner S, Ledbetter DH, Martin CL. 17q12 Recurrent Deletion Syndrome. 2016 Dec 8 [updated 2020 Oct 15]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK401562/ Citation on PubMed
- Moreno-De-Luca D; SGENE Consortium; Mulle JG; Simons Simplex Collection Genetics Consortium; Kaminsky EB, Sanders SJ; GeneSTAR; Myers SM, Adam MP, Pakula AT, Eisenhauer NJ, Uhas K, Weik L, Guy L, Care ME, Morel CF, Boni C, Salbert BA, Chandrareddy A, Demmer LA, Chow EW, Surti U, Aradhya S, Pickering DL, Golden DM, Sanger WG, Aston E, Brothman AR, Gliem TJ, Thorland EC, Ackley T, Iyer R, Huang S, Barber JC, Crolla JA, Warren ST, Martin CL, Ledbetter DH. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet. 2010 Nov 12;87(5):618-30. doi: 10.1016/j.ajhg.2010.10.004. Epub 2010 Nov 4. Erratum In: Am J Hum Genet. 2011 Jan 7;88(1):121. Citation on PubMed or Free article on PubMed Central
- Nagamani SC, Erez A, Shen J, Li C, Roeder E, Cox S, Karaviti L, Pearson M, Kang SH, Sahoo T, Lalani SR, Stankiewicz P, Sutton VR, Cheung SW. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur J Hum Genet. 2010 Mar;18(3):278-84. doi: 10.1038/ejhg.2009.174. Epub 2009 Oct 21. Citation on PubMed or Free article on PubMed Central
- Rasmussen M, Vestergaard EM, Graakjaer J, Petkov Y, Bache I, Fagerberg C, Kibaek M, Svaneby D, Petersen OB, Brasch-Andersen C, Sunde L. 17q12 deletion and duplication syndrome in Denmark-A clinical cohort of 38 patients and review of the literature. Am J Med Genet A. 2016 Nov;170(11):2934-2942. doi: 10.1002/ajmg.a.37848. Epub 2016 Jul 13. Citation on PubMed
- Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, Bjornsdottir G, Walters GB, Jonsdottir GA, Doyle OM, Tost H, Grimm O, Kristjansdottir S, Snorrason H, Davidsdottir SR, Gudmundsson LJ, Jonsson GF, Stefansdottir B, Helgadottir I, Haraldsson M, Jonsdottir B, Thygesen JH, Schwarz AJ, Didriksen M, Stensbol TB, Brammer M, Kapur S, Halldorsson JG, Hreidarsson S, Saemundsen E, Sigurdsson E, Stefansson K. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014 Jan 16;505(7483):361-6. doi: 10.1038/nature12818. Epub 2013 Dec 18. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.