

Description
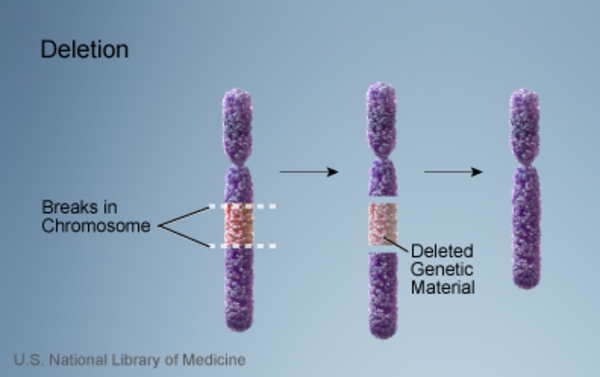
16p12.2 microdeletion is a chromosomal change in which a small amount of genetic material on chromosome 16 is deleted. The deletion occurs on the short (p) arm of the chromosome at a location designated p12.2. Common characteristics that have been described in people with a 16p12.2 microdeletion include developmental delay, delayed speech, intellectual disability that ranges from mild to profound, weak muscle tone (hypotonia), slow growth resulting in short stature, an usually small head (microcephaly), malformations of the heart, recurrent seizures (epilepsy), and psychiatric and behavioral problems.

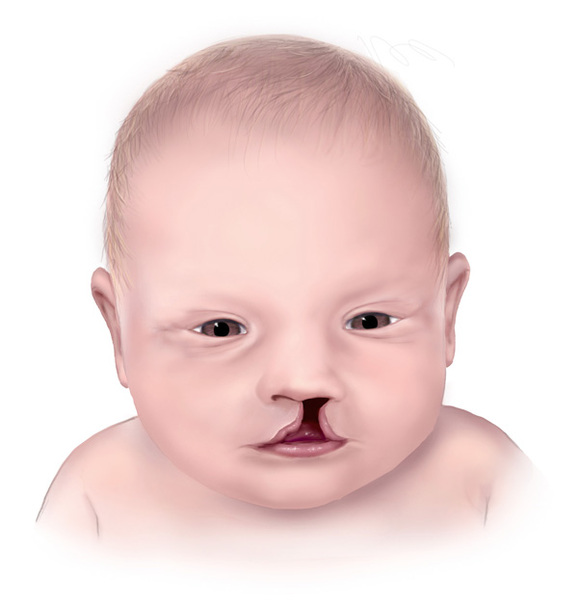
Less common features that can occur in people with a 16p12.2 microdeletion can include hearing loss, an opening in the lip (cleft lip) with or without an opening in the roof of the mouth (cleft palate), dental abnormalities, malformed kidneys, and genital abnormalities in males. However, there is no particular pattern of physical abnormalities that characterizes individuals with a 16p12.2 microdeletion. Signs and symptoms related to the chromosomal change vary even among affected members of the same family, and some people with the deletion have no identified physical or behavioral abnormalities.

Frequency
Researchers estimate that about 1 in 2,000 newborns have a 16p12.2 microdeletion and show signs and symptoms of the condition. However, the actual number may be higher because many people with the microdeletion are likely never diagnosed. Some never come to medical attention because they have no related health or behavioral problems or have only mild signs and symptoms. Others have nonspecific features for which there can be many causes.
Causes
People with a 16p12.2 microdeletion are missing a sequence of about 520,000 DNA building blocks (base pairs), also written as 520 kb, at position p12.2 on chromosome 16. The deleted region contains seven genes and affects one of the two copies of chromosome 16 in each cell.
The signs and symptoms that can result from a 16p12.2 microdeletion are generally related to the loss of one or more genes in this region. However, it is unclear which missing genes contribute to specific features that can occur in the disorder. Because some people with a 16p12.2 microdeletion have no obvious signs or symptoms, researchers believe that other genetic or environmental factors may also be involved. In particular, studies indicate that individuals with a 16p12.2 microdeletion who have neurological or behavioral problems often have an additional, larger chromosomal deletion or duplication on another chromosome. Small duplications of genetic material that occur near the 16p12.2 microdeletion may also contribute to the features associated with this condition.
Inheritance
16p12.2 microdeletion is inherited in an autosomal dominant pattern, which means one copy of the deleted region on chromosome 16 in each cell is sufficient to increase the risk of physical or developmental abnormalities.

In almost all known cases, individuals with a 16p12.2 microdeletion have inherited the chromosomal change from a parent, who may or may not have any related signs or symptoms. The condition is said to have incomplete penetrance because not everyone who has the altered chromosome develops related features.
Other Names for This Condition
- 16p12.1 microdeletion
- Chromosome 16p12.1 deletion syndrome, 520-kb
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Antonacci F, Kidd JM, Marques-Bonet T, Teague B, Ventura M, Girirajan S, Alkan C, Campbell CD, Vives L, Malig M, Rosenfeld JA, Ballif BC, Shaffer LG, Graves TA, Wilson RK, Schwartz DC, Eichler EE. A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat Genet. 2010 Sep;42(9):745-50. doi: 10.1038/ng.643. Epub 2010 Aug 22. Citation on PubMed or Free article on PubMed Central
- Brisset S, Capri Y, Briand-Suleau A, Tosca L, Gras D, Fauret-Amsellem AL, Pineau D, Saada J, Ortonne V, Verloes A, Goossens M, Tachdjian G, Metay C. Inherited 1q21.1q21.2 duplication and 16p11.2 deletion: a two-hit case with more severe clinical manifestations. Eur J Med Genet. 2015 Sep;58(9):497-501. doi: 10.1016/j.ejmg.2015.07.001. Epub 2015 Jul 8. Citation on PubMed
- Coe BP, Girirajan S, Eichler EE. The genetic variability and commonality of neurodevelopmental disease. Am J Med Genet C Semin Med Genet. 2012 May 15;160C(2):118-29. doi: 10.1002/ajmg.c.31327. Epub 2012 Apr 12. Citation on PubMed or Free article on PubMed Central
- Girirajan S, Pizzo L, Moeschler J, Rosenfeld J. 16p12.2 Recurrent Deletion. 2015 Feb 26 [updated 2018 Sep 13]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK274565/ Citation on PubMed
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, Mefford HC, Kidd JM, Browning SR, Browning BL, Dickel DE, Levy DL, Ballif BC, Platky K, Farber DM, Gowans GC, Wetherbee JJ, Asamoah A, Weaver DD, Mark PR, Dickerson J, Garg BP, Ellingwood SA, Smith R, Banks VC, Smith W, McDonald MT, Hoo JJ, French BN, Hudson C, Johnson JP, Ozmore JR, Moeschler JB, Surti U, Escobar LF, El-Khechen D, Gorski JL, Kussmann J, Salbert B, Lacassie Y, Biser A, McDonald-McGinn DM, Zackai EH, Deardorff MA, Shaikh TH, Haan E, Friend KL, Fichera M, Romano C, Gecz J, DeLisi LE, Sebat J, King MC, Shaffer LG, Eichler EE. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010 Mar;42(3):203-9. doi: 10.1038/ng.534. Epub 2010 Feb 14. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.