

Description
Systemic scleroderma is an autoimmune disorder that affects the skin and internal organs. Autoimmune disorders occur when the immune system malfunctions and attacks the body's own tissues and organs. The word "scleroderma" means hard skin in Greek, and the condition is characterized by the buildup of scar tissue (fibrosis) in the skin and other organs. The condition is also called systemic sclerosis because the fibrosis can affect organs other than the skin. Fibrosis is due to the excess production of a tough protein called collagen, which normally strengthens and supports connective tissues throughout the body.
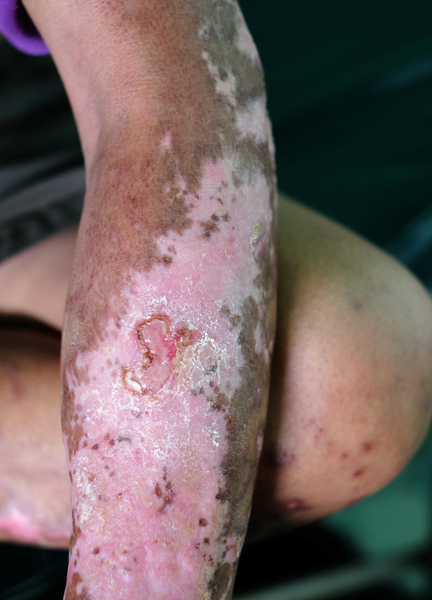
The signs and symptoms of systemic scleroderma usually begin with episodes of Raynaud phenomenon, which can occur weeks to years before fibrosis. In Raynaud phenomenon, the fingers and toes of affected individuals turn white or blue in response to cold temperature or other stresses. This effect occurs because of problems with the small vessels that carry blood to the extremities. Another early sign of systemic scleroderma is puffy or swollen hands before thickening and hardening of the skin due to fibrosis. Skin thickening usually occurs first in the fingers (called sclerodactyly) and may also involve the hands and face. In addition, people with systemic scleroderma often have open sores (ulcers) on their fingers, painful bumps under the skin (calcinosis), or small clusters of enlarged blood vessels just under the skin (telangiectasia).
Fibrosis can also affect internal organs and can lead to impairment or failure of the affected organs. The most commonly affected organs are the esophagus, heart, lungs, and kidneys. Internal organ involvement may be signaled by heartburn, difficulty swallowing (dysphagia), high blood pressure (hypertension), kidney problems, shortness of breath, diarrhea, or impairment of the muscle contractions that move food through the digestive tract (intestinal pseudo-obstruction).
There are three types of systemic scleroderma, defined by the tissues affected in the disorder. In one type of systemic scleroderma, known as limited cutaneous systemic scleroderma, fibrosis usually affects only the hands, arms, and face. Limited cutaneous systemic scleroderma used to be known as CREST syndrome, which is named for the common features of the condition: calcinosis, Raynaud phenomenon, esophageal motility dysfunction, sclerodactyly, and telangiectasia. In another type of systemic scleroderma, known as diffuse cutaneous systemic scleroderma, the fibrosis affects large areas of skin, including the torso and the upper arms and legs, and often involves internal organs. In diffuse cutaneous systemic scleroderma, the condition worsens quickly and organ damage occurs earlier than in other types of the condition. In the third type of systemic scleroderma, called systemic sclerosis sine scleroderma ("sine" means without in Latin), fibrosis affects one or more internal organs but not the skin.
Approximately 15 percent to 25 percent of people with features of systemic scleroderma also have signs and symptoms of another condition that affects connective tissue, such as polymyositis, dermatomyositis, rheumatoid arthritis, Sjögren syndrome, or systemic lupus erythematosus. The combination of systemic scleroderma with other connective tissue abnormalities is known as scleroderma overlap syndrome.
Frequency
The prevalence of systemic scleroderma is estimated to range from 50 to 300 cases per 1 million people. For reasons that are unknown, women are four times more likely to develop the condition than men.
Causes
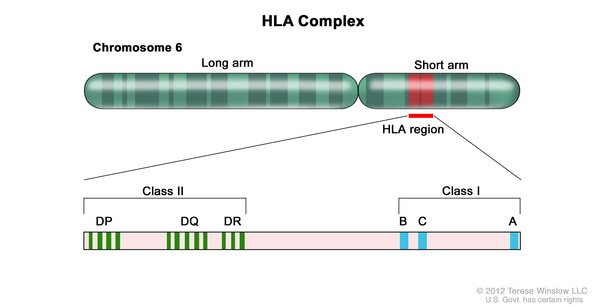
Researchers have identified variations in several genes that may influence the risk of developing systemic scleroderma. The most commonly associated genes belong to a family of genes called the human leukocyte antigen (HLA) complex. The HLA complex helps the immune system distinguish the body's own proteins from proteins made by foreign invaders (such as viruses and bacteria). Each HLA gene has many different normal variations, allowing each person's immune system to react to a wide range of foreign proteins. Specific normal variations of several HLA genes seem to affect the risk of developing systemic scleroderma.
Normal variations in other genes related to the body's immune function, such as IRF5 and STAT4, are also associated with an increased risk of developing systemic scleroderma. Variations in the IRF5 gene are specifically associated with diffuse cutaneous systemic scleroderma, and a variation in the STAT4 gene is associated with limited cutaneous systemic scleroderma. The IRF5 and STAT4 genes both play a role in initiating an immune response when the body detects a foreign invader (pathogen) such as a virus.
It is not known how variations in the associated genes contribute to the increased risk of systemic scleroderma. Variations in multiple genes may work together to increase the risk of developing the condition, and researchers are working to identify and confirm other genes associated with increased risk. In addition, a combination of genetic and environmental factors seems to play a role in developing systemic scleroderma.
Inheritance
Most cases of systemic scleroderma are sporadic, which means they occur in people with no history of the condition in their family. However, some people with systemic scleroderma have close relatives with other autoimmune disorders.
A small percentage of all cases of systemic scleroderma have been reported to run in families; however, the condition does not have a clear pattern of inheritance. Multiple genetic and environmental factors likely play a part in determining the risk of developing this condition. As a result, inheriting a genetic variation linked with systemic scleroderma does not mean that a person will develop the condition.
Other Names for This Condition
- Familial progressive scleroderma
- Progressive scleroderma
- Systemic sclerosis
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Allanore Y, Saad M, Dieude P, Avouac J, Distler JH, Amouyel P, Matucci-Cerinic M, Riemekasten G, Airo P, Melchers I, Hachulla E, Cusi D, Wichmann HE, Wipff J, Lambert JC, Hunzelmann N, Tiev K, Caramaschi P, Diot E, Kowal-Bielecka O, Valentini G, Mouthon L, Czirjak L, Damjanov N, Salvi E, Conti C, Muller M, Muller-Ladner U, Riccieri V, Ruiz B, Cracowski JL, Letenneur L, Dupuy AM, Meyer O, Kahan A, Munnich A, Boileau C, Martinez M. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011 Jul;7(7):e1002091. doi: 10.1371/journal.pgen.1002091. Epub 2011 Jul 7. Citation on PubMed or Free article on PubMed Central
- Dieude P, Guedj M, Wipff J, Avouac J, Fajardy I, Diot E, Granel B, Sibilia J, Cabane J, Mouthon L, Cracowski JL, Carpentier PH, Hachulla E, Meyer O, Kahan A, Boileau C, Allanore Y. Association between the IRF5 rs2004640 functional polymorphism and systemic sclerosis: a new perspective for pulmonary fibrosis. Arthritis Rheum. 2009 Jan;60(1):225-33. doi: 10.1002/art.24183. Citation on PubMed
- Hachulla E, Launay D. Diagnosis and classification of systemic sclerosis. Clin Rev Allergy Immunol. 2011 Apr;40(2):78-83. doi: 10.1007/s12016-010-8198-y. Citation on PubMed
- Hua-Huy T, Dinh-Xuan AT. Cellular and molecular mechanisms in the pathophysiology of systemic sclerosis. Pathol Biol (Paris). 2015 Apr;63(2):61-8. doi: 10.1016/j.patbio.2015.03.003. Epub 2015 Mar 25. Citation on PubMed
- Ito I, Kawaguchi Y, Kawasaki A, Hasegawa M, Ohashi J, Hikami K, Kawamoto M, Fujimoto M, Takehara K, Sato S, Hara M, Tsuchiya N. Association of a functional polymorphism in the IRF5 region with systemic sclerosis in a Japanese population. Arthritis Rheum. 2009 Jun;60(6):1845-50. doi: 10.1002/art.24600. Citation on PubMed
- Korman BD, Criswell LA. Recent advances in the genetics of systemic sclerosis: toward biological and clinical significance. Curr Rheumatol Rep. 2015 Mar;17(3):21. doi: 10.1007/s11926-014-0484-x. Citation on PubMed or Free article on PubMed Central
- Rueda B, Broen J, Simeon C, Hesselstrand R, Diaz B, Suarez H, Ortego-Centeno N, Riemekasten G, Fonollosa V, Vonk MC, van den Hoogen FH, Sanchez-Roman J, Aguirre-Zamorano MA, Garcia-Portales R, Pros A, Camps MT, Gonzalez-Gay MA, Coenen MJ, Airo P, Beretta L, Scorza R, van Laar J, Gonzalez-Escribano MF, Nelson JL, Radstake TR, Martin J. The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum Mol Genet. 2009 Jun 1;18(11):2071-7. doi: 10.1093/hmg/ddp119. Epub 2009 Mar 13. Citation on PubMed
- Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, Kaplan MH. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity. 2008 Nov 14;29(5):679-90. doi: 10.1016/j.immuni.2008.08.017. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.