

Description
Retinoblastoma is a rare type of eye cancer that usually develops in early childhood, typically before the age of 5. This form of cancer develops in the retina, which is the specialized light-sensitive tissue at the back of the eye that detects light and color.

In children with retinoblastoma, the disease often affects only one eye. However, one out of three children with retinoblastoma develops cancer in both eyes. The most common first sign of retinoblastoma is a visible whiteness in the pupil called "cat's eye reflex" or leukocoria. This unusual whiteness is particularly noticeable in dim light or in photographs taken with a flash. Other signs and symptoms of retinoblastoma include crossed eyes or eyes that do not point in the same direction (strabismus), which can cause squinting; a change in the color of the colored part of the eye (iris); redness, soreness, or swelling of the eyelids; and blindness or poor vision in the affected eye or eyes.


Retinoblastoma is often curable when it is diagnosed early. However, if it is not treated promptly, this cancer can spread beyond the eye to other parts of the body. This advanced form of retinoblastoma can be life-threatening.
When retinoblastoma is associated with a genetic change (mutation) that occurs in all of the body's cells, it is known as hereditary (or germinal) retinoblastoma. People with this form of retinoblastoma typically develop cancer in both eyes and also have an increased risk of developing several other cancers outside the eye. Specifically, they are more likely to develop a cancer of the pineal gland in the brain (pineoblastoma), a type of bone cancer known as osteosarcoma, cancers of soft tissues (such as muscle) called soft tissue sarcomas, and an aggressive form of skin cancer called melanoma.
Frequency
Retinoblastoma is diagnosed in 250 to 350 children per year in the United States. It accounts for about 4 percent of all cancers in children younger than 15 years.
Causes
Mutations in the RB1 gene are responsible for most cases of retinoblastoma. RB1 is a tumor suppressor gene, which means that it normally regulates cell growth and stops cells from dividing too rapidly or in an uncontrolled way. Most mutations in the RB1 gene prevent it from making any functional protein, so cells are unable to regulate cell division effectively. As a result, certain cells in the retina can divide uncontrollably to form a cancerous tumor. Some studies suggest that additional genetic changes can influence the development of retinoblastoma; these changes may help explain variations in the development and growth of retinoblastoma and other types of tumors in different people.
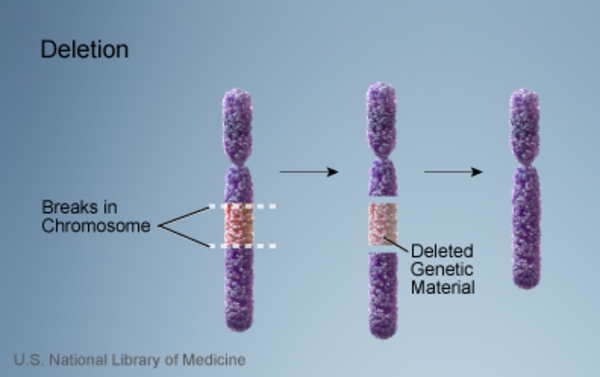
A small percentage of retinoblastomas are caused by deletions in the region of chromosome 13 that contains the RB1 gene. Because these chromosomal changes involve several genes in addition to RB1, affected children usually also have intellectual disability, slow growth, and distinctive facial features (such as prominent eyebrows, a short nose with a broad nasal bridge, and ear abnormalities).
Inheritance
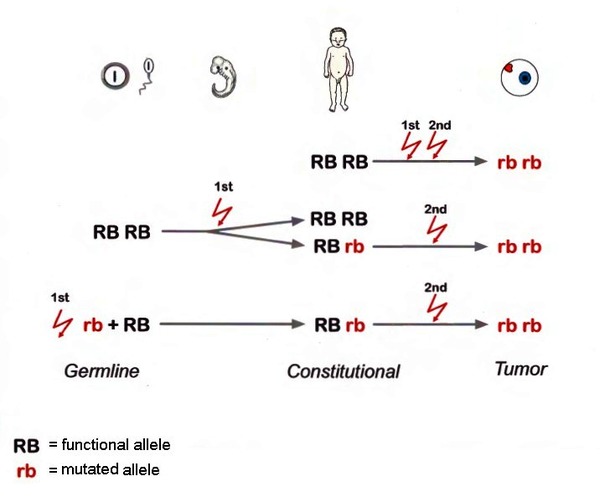
Researchers estimate that one-third of all retinoblastomas are hereditary, which means that RB1 gene mutations are present in all of the body's cells, including reproductive cells (sperm or eggs). People with hereditary retinoblastoma may have a family history of the disease, and they are at risk of passing on the mutated RB1 gene to the next generation. The other two-thirds of retinoblastomas are non-hereditary, which means that RB1 gene mutations are present only in cells of the eye and cannot be passed to the next generation.
In hereditary retinoblastoma, mutations in the RB1 gene appear to be inherited in an autosomal dominant pattern. Autosomal dominant inheritance means that one copy of the altered gene in each cell is sufficient to increase the risk of cancer. A person with hereditary retinoblastoma may inherit an altered copy of the RB1 gene from one parent, or the altered gene may be the result of a new mutation that occurs in an egg or sperm cell or just after fertilization. For retinoblastoma to develop, a mutation involving the other copy of the RB1 gene must occur in retinal cells during the person's lifetime. This second mutation usually occurs in childhood, typically leading to the development of retinoblastoma in both eyes.


In the non-hereditary form of retinoblastoma, typically only one eye is affected and there is no family history of the disease. Affected individuals are born with two normal copies of the RB1 gene. Then, usually in early childhood, both copies of the RB1 gene in certain retinal cells acquire mutations. People with non-hereditary retinoblastoma are not at risk of passing these RB1 gene mutations to their children. However, without genetic testing it can be difficult to tell whether a person with retinoblastoma in one eye has the hereditary or the non-hereditary form of the disease.
Other Names for This Condition
- Glioma, retinal
- RB
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Baud O, Cormier-Daire V, Lyonnet S, Desjardins L, Turleau C, Doz F. Dysmorphic phenotype and neurological impairment in 22 retinoblastoma patients with constitutional cytogenetic 13q deletion. Clin Genet. 1999 Jun;55(6):478-82. doi: 10.1034/j.1399-0004.1999.550614.x. Citation on PubMed
- Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007 Jul;46(7):617-34. doi: 10.1002/gcc.20457. Citation on PubMed
- De Falco G, Giordano A. pRb2/p130: a new candidate for retinoblastoma tumor formation. Oncogene. 2006 Aug 28;25(38):5333-40. doi: 10.1038/sj.onc.1209614. Citation on PubMed
- Ewens KG, Bhatti TR, Moran KA, Richards-Yutz J, Shields CL, Eagle RC, Ganguly A. Phosphorylation of pRb: mechanism for RB pathway inactivation in MYCN-amplified retinoblastoma. Cancer Med. 2017 Mar;6(3):619-630. doi: 10.1002/cam4.1010. Epub 2017 Feb 17. Citation on PubMed or Free article on PubMed Central
- Lohmann DR, Gallie BL. Retinoblastoma. 2000 Jul 18 [updated 2023 Sep 21]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1452/ Citation on PubMed
- Madhavan J, Ganesh A, Kumaramanickavel G. Retinoblastoma: from disease to discovery. Ophthalmic Res. 2008;40(5):221-6. doi: 10.1159/000128578. Epub 2008 Apr 29. Citation on PubMed
- Mallipatna A, Marino M, Singh AD. Genetics of Retinoblastoma. Asia Pac J Ophthalmol (Phila). 2016 Jul-Aug;5(4):260-4. doi: 10.1097/APO.0000000000000219. Citation on PubMed
- Poulaki V, Mukai S. Retinoblastoma: genetics and pathology. Int Ophthalmol Clin. 2009 Winter;49(1):155-64. doi: 10.1097/IIO.0b013e3181924bc2. No abstract available. Citation on PubMed
- Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Theriault BL, Prigoda-Lee NL, Spencer C, Dimaras H, Corson TW, Pang R, Massey C, Godbout R, Jiang Z, Zacksenhaus E, Paton K, Moll AC, Houdayer C, Raizis A, Halliday W, Lam WL, Boutros PC, Lohmann D, Dorsman JC, Gallie BL. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013 Apr;14(4):327-34. doi: 10.1016/S1470-2045(13)70045-7. Epub 2013 Mar 13. Citation on PubMed
- Schefler AC, Abramson DH. Retinoblastoma: what is new in 2007-2008. Curr Opin Ophthalmol. 2008 Nov;19(6):526-34. doi: 10.1097/ICU.0b013e328312975b. Citation on PubMed
- Sippel KC, Fraioli RE, Smith GD, Schalkoff ME, Sutherland J, Gallie BL, Dryja TP. Frequency of somatic and germ-line mosaicism in retinoblastoma: implications for genetic counseling. Am J Hum Genet. 1998 Mar;62(3):610-9. doi: 10.1086/301766. Citation on PubMed or Free article on PubMed Central
- Soliman SE, Dimaras H, Khetan V, Gardiner JA, Chan HS, Heon E, Gallie BL. Prenatal versus Postnatal Screening for Familial Retinoblastoma. Ophthalmology. 2016 Dec;123(12):2610-2617. doi: 10.1016/j.ophtha.2016.08.027. Epub 2016 Oct 3. Citation on PubMed
- Soliman SE, Racher H, Zhang C, MacDonald H, Gallie BL. Genetics and Molecular Diagnostics in Retinoblastoma--An Update. Asia Pac J Ophthalmol (Phila). 2017 Mar-Apr;6(2):197-207. doi: 10.22608/APO.201711. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.