

Description
The Say-Barber-Biesecker-Young-Simpson (SBBYS) variant of Ohdo syndrome is a rare condition characterized by genital abnormalities in males, missing or underdeveloped kneecaps (patellae), intellectual disability, distinctive facial features, and abnormalities affecting other parts of the body.
Males with the SBBYS variant of Ohdo syndrome typically have undescended testes (cryptorchidism). Females with this condition have normal genitalia.
Missing or underdeveloped patellae is the most common skeletal abnormality associated with the SBBYS variant of Ohdo syndrome. Affected individuals also have joint stiffness involving the hips, knees, and ankles that can impair movement. Although joints in the lower body are stiff, joints in the arms and upper body may be unusually loose (lax). Many people with this condition have long thumbs and first (big) toes.
The SBBYS variant of Ohdo syndrome is also associated with delayed development and intellectual disability, which are often severe. Many affected infants have weak muscle tone (hypotonia) that leads to breathing and feeding difficulties.
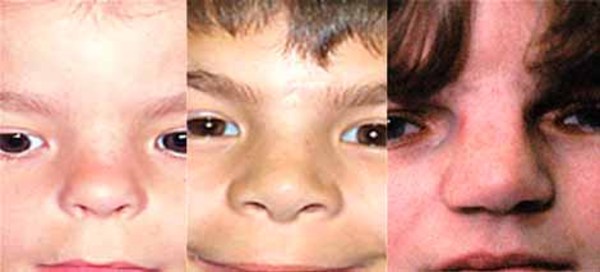
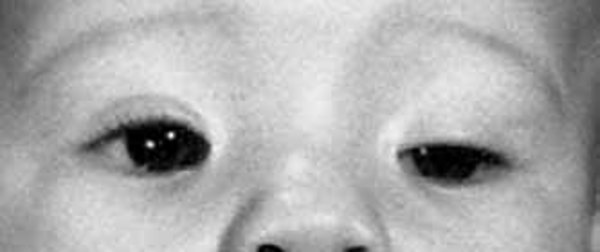
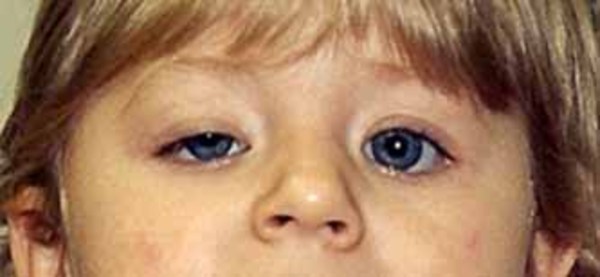
The SBBYS variant of Ohdo syndrome is characterized by a mask-like, non-expressive face. Additionally, affected individuals may have distinctive facial features such as prominent cheeks, a broad nasal bridge or a nose with a rounded tip, a narrowing of the eye opening (blepharophimosis), droopy eyelids (ptosis), and abnormalities of the tear (lacrimal) glands. About one-third of affected individuals are born with an opening in the roof of the mouth called a cleft palate. The SBBYS variant of Ohdo syndrome can also be associated with heart defects and dental problems.

Frequency
The SBBYS variant of Ohdo syndrome is estimated to occur in fewer than 1 per million people. At least 19 cases have been reported in the medical literature.
Causes
The SBBYS variant of Ohdo syndrome is caused by mutations in the KAT6B gene. This gene provides instructions for making a type of enzyme called a histone acetyltransferase. These enzymes modify histones, which are structural proteins that attach (bind) to DNA and give chromosomes their shape. By adding a small molecule called an acetyl group to histones, histone acetyltransferases control the activity of certain genes. Little is known about the function of the histone acetyltransferase produced from the KAT6B gene. It appears to regulate genes that are important for early development, including development of the skeleton and nervous system.
The mutations that cause the SBBYS variant of Ohdo syndrome likely prevent the production of functional histone acetyltransferase from one copy of the KAT6B gene in each cell. Studies suggest that the resulting shortage of this enzyme impairs the regulation of various genes during early development. However, it is unclear how these changes lead to the specific features of the condition.
Inheritance
This condition has an autosomal dominant inheritance pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Almost all reported cases have resulted from new mutations in the gene and have occurred in people with no history of the disorder in their family.

Other Names for This Condition
- Blepharophimosis and mental retardation syndrome, Say-Barber/Biesecker/Young-Simpson type
- Blepharophimosis-intellectual deficit syndrome, Say-Barber/Biesecker/Young-Simpson type
- BMRS SBBYS
- Ohdo syndrome, Say-Barber-Biesecker variant
- Ohdo syndrome, SBBYS variant
- Say-Barber-Biesecker-Young-Simpson syndrome
- Say-Barber-Biesecker-Young-Simpson variant of Ohdo syndrome
- SBBYS variant of Ohdo syndrome
- SBBYSS
- Young-Simpson syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Campeau PM, Lu JT, Dawson BC, Fokkema IF, Robertson SP, Gibbs RA, Lee BH. The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum Mutat. 2012 Nov;33(11):1520-5. doi: 10.1002/humu.22141. Epub 2012 Jul 12. Citation on PubMed or Free article on PubMed Central
- Clayton-Smith J, O'Sullivan J, Daly S, Bhaskar S, Day R, Anderson B, Voss AK, Thomas T, Biesecker LG, Smith P, Fryer A, Chandler KE, Kerr B, Tassabehji M, Lynch SA, Krajewska-Walasek M, McKee S, Smith J, Sweeney E, Mansour S, Mohammed S, Donnai D, Black G. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011 Nov 11;89(5):675-81. doi: 10.1016/j.ajhg.2011.10.008. Citation on PubMed or Free article on PubMed Central
- Day R, Beckett B, Donnai D, Fryer A, Heidenblad M, Howard P, Kerr B, Mansour S, Maye U, McKee S, Mohammed S, Sweeney E, Tassabehji M, de Vries BB, Clayton-Smith J. A clinical and genetic study of the Say/Barber/Biesecker/Young-Simpson type of Ohdo syndrome. Clin Genet. 2008 Nov;74(5):434-44. doi: 10.1111/j.1399-0004.2008.01087.x. Epub 2008 Sep 16. Citation on PubMed
- Lemire G, Campeau PM, Lee BH. KAT6B Disorders. 2012 Dec 13 [updated 2020 Jan 2]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK114806/ Citation on PubMed
- Masuno M, Imaizumi K, Okada T, Adachi M, Nishimura G, Ishii T, Tachibana K, Kuroki Y. Young-Simpson syndrome: further delineation of a distinct syndrome with congenital hypothyroidism, congenital heart defects, facial dysmorphism, and mental retardation. Am J Med Genet. 1999 May 7;84(1):8-11. doi: 10.1002/(sici)1096-8628(19990507)84:13.0.co;2-2. Citation on PubMed
- Verloes A, Bremond-Gignac D, Isidor B, David A, Baumann C, Leroy MA, Stevens R, Gillerot Y, Heron D, Heron B, Benzacken B, Lacombe D, Brunner H, Bitoun P. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A. 2006 Jun 15;140(12):1285-96. doi: 10.1002/ajmg.a.31270. Citation on PubMed
- White SM, Ades LC, Amor D, Liebelt J, Bankier A, Baker E, Wilson M, Savarirayan R. Two further cases of Ohdo syndrome delineate the phenotypic variability of the condition. Clin Dysmorphol. 2003 Apr;12(2):109-13. doi: 10.1097/00019605-200304000-00007. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.