

Description
Marfan syndrome is a disorder that affects the connective tissue in many parts of the body. Connective tissue provides strength and flexibility to structures such as bones, ligaments, muscles, blood vessels, and heart valves. The signs and symptoms of Marfan syndrome vary widely in severity, timing of onset, and rate of progression.
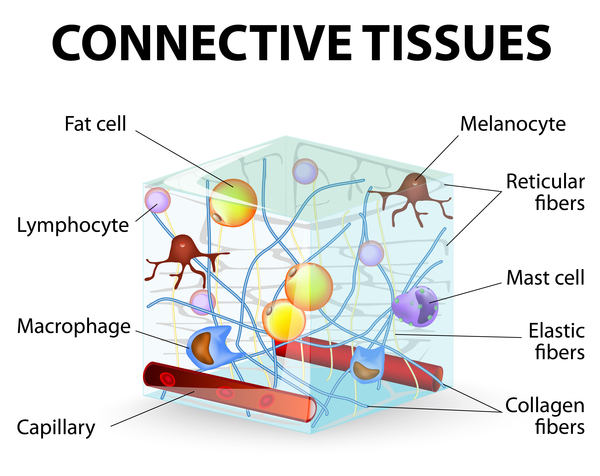
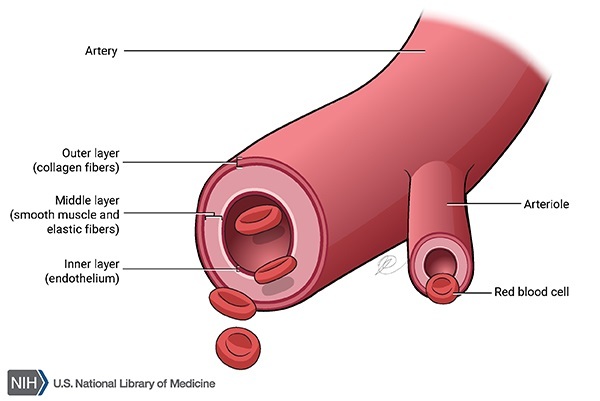
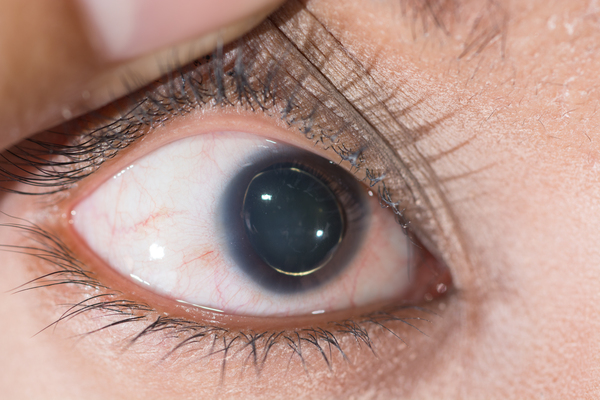
Because connective tissue is found throughout the body, Marfan syndrome can affect many systems, often causing abnormalities in the heart, blood vessels, eyes, bones, and joints. The two primary features of Marfan syndrome are vision problems caused by a dislocated lens (ectopia lentis) in one or both eyes and defects in the large blood vessel that distributes blood from the heart to the rest of the body (the aorta). The aorta can weaken and stretch, which may lead to a bulge in the blood vessel wall (an aneurysm). Stretching of the aorta may cause the aortic valve to leak, which can lead to a sudden tearing of the layers in the aorta wall (aortic dissection). Aortic aneurysm and dissection can be life threatening.

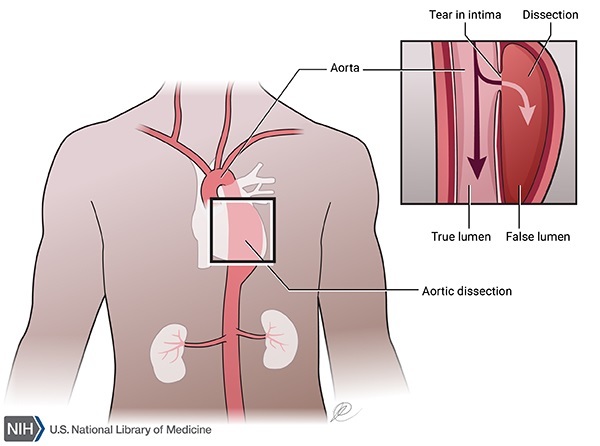
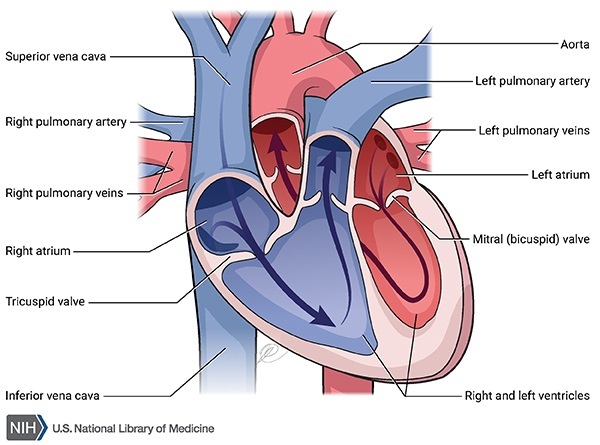
Many people with Marfan syndrome have additional heart problems including a leak in the valve that connects two of the four chambers of the heart (mitral valve prolapse) or the valve that regulates blood flow from the heart into the aorta (aortic valve regurgitation). Leaks in these valves can cause shortness of breath, fatigue, and an irregular heartbeat felt as skipped or extra beats (palpitations).
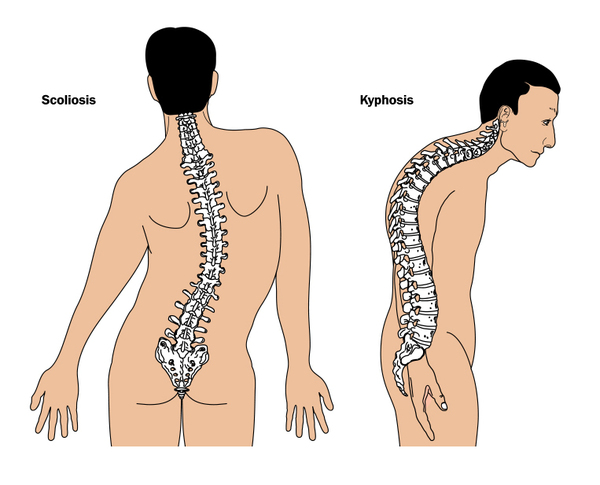
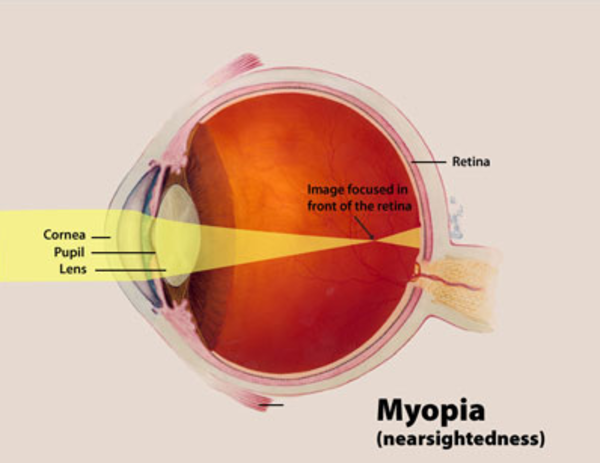
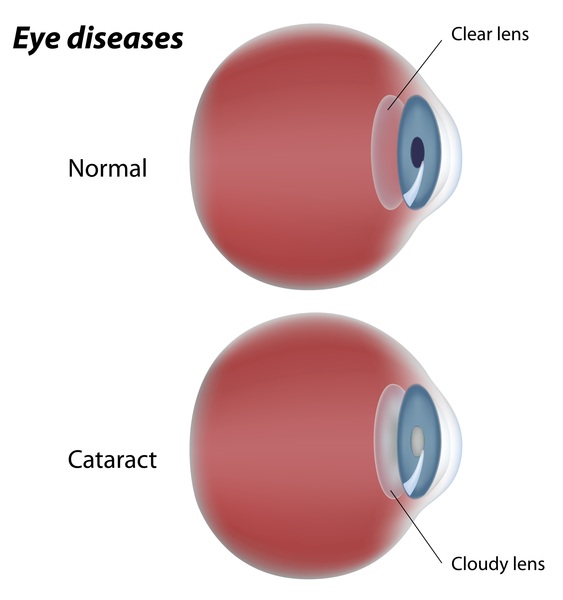
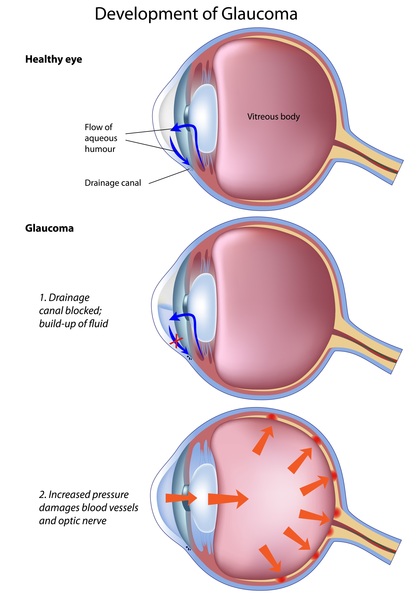
Individuals with Marfan syndrome are usually tall and slender, have elongated fingers and toes (arachnodactyly), loose joints, and have an arm span that exceeds their body height. Other common features include a long and narrow face, crowded teeth, an abnormal curvature of the spine (scoliosis or kyphosis), stretch marks (striae) not related to weight gain or loss, and either a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum). Some individuals develop an abnormal accumulation of air in the chest cavity that can result in the collapse of a lung (spontaneous pneumothorax). A membrane called the dura, which surrounds the brain and spinal cord, can be abnormally enlarged (dural ectasia) in people with Marfan syndrome. Dural ectasia can cause pain in the back, abdomen, legs, or head. Most individuals with Marfan syndrome have some degree of nearsightedness (myopia). Clouding of the lens (cataract) may occur in mid-adulthood, and increased pressure within the eye (glaucoma) occurs more frequently in people with Marfan syndrome than in those without the condition.
The features of Marfan syndrome can become apparent anytime between infancy and adulthood. Depending on the onset and severity of signs and symptoms, Marfan syndrome can be fatal early in life; however, with proper treatment, many affected individuals have normal lifespans.
Frequency
The incidence of Marfan syndrome is approximately 1 in 5,000 worldwide.
Causes
Mutations in the FBN1 gene cause Marfan syndrome. The FBN1 gene provides instructions for making a protein called fibrillin-1. Fibrillin-1 attaches (binds) to other fibrillin-1 proteins and other molecules to form threadlike filaments called microfibrils. Microfibrils become part of the fibers that provide strength and flexibility to connective tissue. Additionally, microfibrils bind to molecules called growth factors and release them at various times to control the growth and repair of tissues and organs throughout the body. A mutation in the FBN1 gene can reduce the amount of functional fibrillin-1 that is available to form microfibrils, which leads to decreased microfibril formation. As a result, microfibrils cannot bind to growth factors, so excess growth factors are available and elasticity in many tissues is decreased, leading to overgrowth and instability of tissues in Marfan syndrome.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

At least 25 percent of Marfan syndrome cases result from a new mutation in the FBN1 gene. These cases occur in people with no history of the disorder in their family.

Other Names for This Condition
- Marfan's syndrome
- MFS
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Canadas V, Vilacosta I, Bruna I, Fuster V. Marfan syndrome. Part 1: pathophysiology and diagnosis. Nat Rev Cardiol. 2010 May;7(5):256-65. doi: 10.1038/nrcardio.2010.30. Epub 2010 Mar 30. Citation on PubMed
- Chaffins JA. Marfan syndrome. Radiol Technol. 2007 Jan-Feb;78(3):222-36; quiz 237-9. Citation on PubMed
- Faivre L, Masurel-Paulet A, Collod-Beroud G, Callewaert BL, Child AH, Stheneur C, Binquet C, Gautier E, Chevallier B, Huet F, Loeys BL, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Grasso M, Halliday DJ, Beroud C, Bonithon-Kopp C, Claustres M, Robinson PN, Ades L, De Backer J, Coucke P, Francke U, De Paepe A, Boileau C, Jondeau G. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics. 2009 Jan;123(1):391-8. doi: 10.1542/peds.2008-0703. Citation on PubMed
- Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation. 2008 May 27;117(21):2802-13. doi: 10.1161/CIRCULATIONAHA.107.693523. No abstract available. Citation on PubMed
- Kirschner R, Hubmacher D, Iyengar G, Kaur J, Fagotto-Kaufmann C, Bromme D, Bartels R, Reinhardt DP. Classical and neonatal Marfan syndrome mutations in fibrillin-1 cause differential protease susceptibilities and protein function. J Biol Chem. 2011 Sep 16;286(37):32810-23. doi: 10.1074/jbc.M111.221804. Epub 2011 Jul 22. Citation on PubMed or Free article on PubMed Central
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010 Jul;47(7):476-85. doi: 10.1136/jmg.2009.072785. Citation on PubMed
- Mizuguchi T, Matsumoto N. Recent progress in genetics of Marfan syndrome and Marfan-associated disorders. J Hum Genet. 2007;52(1):1-12. doi: 10.1007/s10038-006-0078-1. Epub 2006 Oct 24. Citation on PubMed
- National Institute of Arthritis and Musculoskeletal and Skin Diseases
- Pearson GD, Devereux R, Loeys B, Maslen C, Milewicz D, Pyeritz R, Ramirez F, Rifkin D, Sakai L, Svensson L, Wessels A, Van Eyk J, Dietz HC; National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group. Report of the National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group on research in Marfan syndrome and related disorders. Circulation. 2008 Aug 12;118(7):785-91. doi: 10.1161/CIRCULATIONAHA.108.783753. No abstract available. Citation on PubMed or Free article on PubMed Central
- Pyeritz RE, Loeys B. The 8th international research symposium on the Marfan syndrome and related conditions. Am J Med Genet A. 2012 Jan;158A(1):42-9. doi: 10.1002/ajmg.a.34386. Epub 2011 Dec 2. Citation on PubMed
- Pyeritz RE; American College of Medical Genetics and Genomics. Evaluation of the adolescent or adult with some features of Marfan syndrome. Genet Med. 2012 Jan;14(1):171-7. doi: 10.1038/gim.2011.48. Epub 2012 Jan 5. Citation on PubMed
- Robinson PN, Arteaga-Solis E, Baldock C, Collod-Beroud G, Booms P, De Paepe A, Dietz HC, Guo G, Handford PA, Judge DP, Kielty CM, Loeys B, Milewicz DM, Ney A, Ramirez F, Reinhardt DP, Tiedemann K, Whiteman P, Godfrey M. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006 Oct;43(10):769-87. doi: 10.1136/jmg.2005.039669. Epub 2006 Mar 29. Citation on PubMed or Free article on PubMed Central
- Singh KK, Rommel K, Mishra A, Karck M, Haverich A, Schmidtke J, Arslan-Kirchner M. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum Mutat. 2006 Aug;27(8):770-7. doi: 10.1002/humu.20354. Citation on PubMed
- The Marfan Foundation
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.