

Description
Fanconi anemia is a condition that affects many parts of the body. People with this condition may have bone marrow failure, physical abnormalities, organ defects, and an increased risk of certain cancers.
The major function of bone marrow is to produce new blood cells. These include red blood cells, which carry oxygen to the body's tissues; white blood cells, which fight infections; and platelets, which are necessary for normal blood clotting. Approximately 90 percent of people with Fanconi anemia have impaired bone marrow function that leads to a decrease in the production of all blood cells (aplastic anemia). Affected individuals experience extreme tiredness (fatigue) due to low numbers of red blood cells (anemia), frequent infections due to low numbers of white blood cells (neutropenia), and clotting problems due to low numbers of platelets (thrombocytopenia). People with Fanconi anemia may also develop myelodysplastic syndrome, a condition in which immature blood cells fail to develop normally.
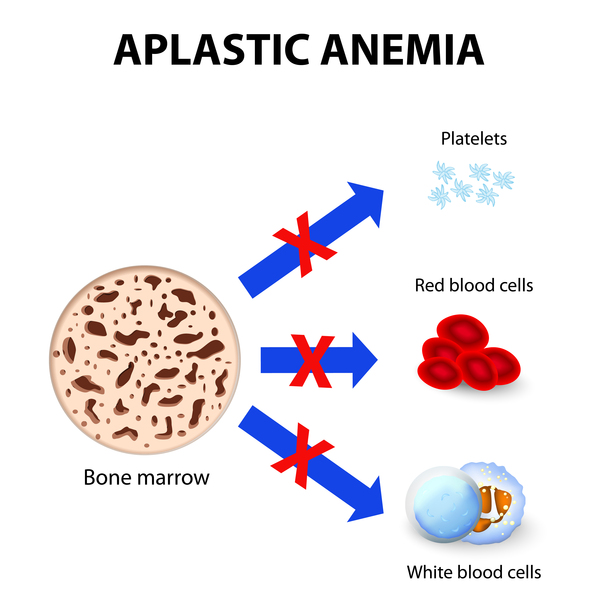

More than half of people with Fanconi anemia have physical abnormalities. These abnormalities can involve irregular skin coloring such as unusually light-colored skin (hypopigmentation) or café-au-lait spots, which are flat patches on the skin that are darker than the surrounding area. Other possible symptoms of Fanconi anemia include malformed thumbs or forearms and other skeletal problems including short stature; malformed or absent kidneys and other defects of the urinary tract; gastrointestinal abnormalities; heart defects; eye abnormalities such as small or abnormally shaped eyes; and malformed ears and hearing loss. People with this condition may have abnormal genitalia or malformations of the reproductive system. As a result, most affected males and about half of affected females cannot have biological children (are infertile). Additional signs and symptoms can include abnormalities of the brain and spinal cord (central nervous system), including increased fluid in the center of the brain (hydrocephalus) or an unusually small head size (microcephaly).


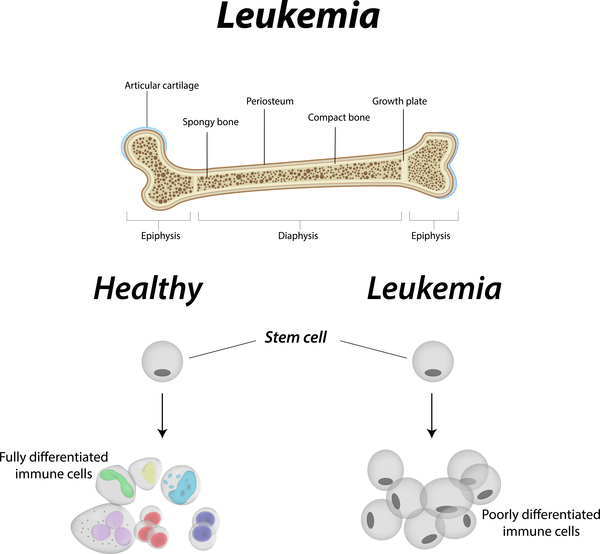
Individuals with Fanconi anemia have an increased risk of developing a cancer of blood-forming cells in the bone marrow called acute myeloid leukemia (AML) or tumors of the head, neck, skin, gastrointestinal system, or genital tract. The likelihood of developing one of these cancers in people with Fanconi anemia is between 10 and 30 percent.
Frequency
Fanconi anemia occurs in 1 in 160,000 individuals worldwide. This condition is more common among people of Ashkenazi Jewish descent, the Roma population of Spain, and Black South Africans.
Causes
Mutations in at least 15 genes can cause Fanconi anemia. Proteins produced from these genes are involved in a cell process known as the FA pathway. The FA pathway is turned on (activated) when the process of making new copies of DNA, called DNA replication, is blocked due to DNA damage. The FA pathway sends certain proteins to the area of damage, which trigger DNA repair so DNA replication can continue.
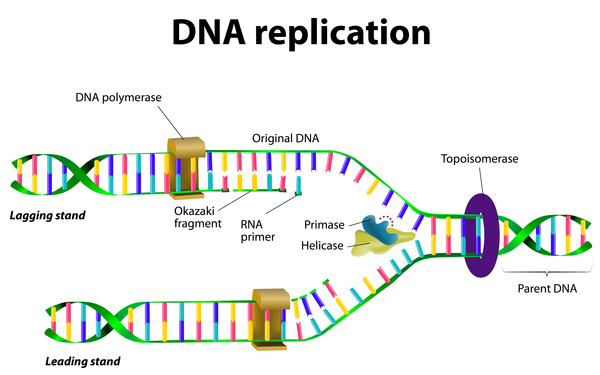
The FA pathway is particularly responsive to a certain type of DNA damage known as interstrand cross-links (ICLs). ICLs occur when two DNA building blocks (nucleotides) on opposite strands of DNA are abnormally attached or linked together, which stops the process of DNA replication. ICLs can be caused by a buildup of toxic substances produced in the body or by treatment with certain cancer therapy drugs.
Eight proteins associated with Fanconi anemia group together to form a complex known as the FA core complex. The FA core complex activates two proteins, called FANCD2 and FANCI. The activation of these two proteins brings DNA repair proteins to the area of the ICL so the cross-link can be removed and DNA replication can continue.
Eighty to 90 percent of cases of Fanconi anemia are due to mutations in one of three genes, FANCA, FANCC, and FANCG. These genes provide instructions for producing components of the FA core complex. Mutations in any of the many genes associated with the FA core complex will cause the complex to be nonfunctional and disrupt the entire FA pathway. As a result, DNA damage is not repaired efficiently and ICLs build up over time. The ICLs stall DNA replication, ultimately resulting in either abnormal cell death due to an inability make new DNA molecules or uncontrolled cell growth due to a lack of DNA repair processes. Cells that divide quickly, such as bone marrow cells and cells of the developing fetus, are particularly affected. The death of these cells results in the decrease in blood cells and the physical abnormalities characteristic of Fanconi anemia. When the buildup of errors in DNA leads to uncontrolled cell growth, affected individuals can develop acute myeloid leukemia or other cancers.

Inheritance
Fanconi anemia is most often inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Very rarely, this condition is inherited in an X-linked recessive pattern. The gene associated with X-linked recessive Fanconi anemia is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

Other Names for This Condition
- FA
- Fanconi hypoplastic anemia
- Fanconi pancytopenia
- Fanconi panmyelopathy
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Fanconi anemia

- Genetic Testing Registry: Fanconi anemia, complementation group M
- Genetic Testing Registry: Fanconi anemia complementation group A
- Genetic Testing Registry: Fanconi anemia complementation group B
- Genetic Testing Registry: Fanconi anemia complementation group C
- Genetic Testing Registry: Fanconi anemia complementation group D1
- Genetic Testing Registry: Fanconi anemia complementation group D2
- Genetic Testing Registry: Fanconi anemia complementation group E
- Genetic Testing Registry: Fanconi anemia complementation group F
- Genetic Testing Registry: Fanconi anemia complementation group G
- Genetic Testing Registry: Fanconi anemia complementation group I
- Genetic Testing Registry: Fanconi anemia complementation group J
- Genetic Testing Registry: Fanconi anemia complementation group L
- Genetic Testing Registry: Fanconi anemia complementation group N
- Genetic Testing Registry: Fanconi anemia complementation group O
- Genetic Testing Registry: Fanconi anemia complementation group P
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- FANCONI ANEMIA, COMPLEMENTATION GROUP E; FANCE
- FANCONI ANEMIA, COMPLEMENTATION GROUP B; FANCB
- FANCONI ANEMIA, COMPLEMENTATION GROUP C; FANCC
- FANCONI ANEMIA, COMPLEMENTATION GROUP D2; FANCD2
- FANCONI ANEMIA, COMPLEMENTATION GROUP A; FANCA
- FANCONI ANEMIA, COMPLEMENTATION GROUP F; FANCF
- FANCONI ANEMIA, COMPLEMENTATION GROUP D1; FANCD1
- FANCONI ANEMIA, COMPLEMENTATION GROUP I; FANCI
- FANCONI ANEMIA, COMPLEMENTATION GROUP J; FANCJ
- FANCONI ANEMIA, COMPLEMENTATION GROUP N; FANCN
- FANCM GENE; FANCM
- FANCONI ANEMIA, COMPLEMENTATION GROUP G; FANCG
- FANCONI ANEMIA, COMPLEMENTATION GROUP L; FANCL
- FANCONI ANEMIA, COMPLEMENTATION GROUP O; FANCO
- FANCONI ANEMIA, COMPLEMENTATION GROUP P; FANCP
Scientific Articles on PubMed
References
- Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009 Jul 31;668(1-2):4-10. doi: 10.1016/j.mrfmmm.2009.01.013. Epub 2009 Feb 28. Citation on PubMed or Free article on PubMed Central
- de Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutat Res. 2009 Jul 31;668(1-2):11-9. doi: 10.1016/j.mrfmmm.2008.11.004. Epub 2008 Nov 14. Citation on PubMed
- Deakyne JS, Mazin AV. Fanconi anemia: at the crossroads of DNA repair. Biochemistry (Mosc). 2011 Jan;76(1):36-48. doi: 10.1134/s0006297911010068. Citation on PubMed
- Green AM, Kupfer GM. Fanconi anemia. Hematol Oncol Clin North Am. 2009 Apr;23(2):193-214. doi: 10.1016/j.hoc.2009.01.008. Citation on PubMed
- Kee Y, D'Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010 Aug 15;24(16):1680-94. doi: 10.1101/gad.1955310. Citation on PubMed or Free article on PubMed Central
- Kitao H, Takata M. Fanconi anemia: a disorder defective in the DNA damage response. Int J Hematol. 2011 Apr;93(4):417-424. doi: 10.1007/s12185-011-0777-z. Epub 2011 Feb 18. Citation on PubMed
- Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006 Sep 25;25(43):5875-84. doi: 10.1038/sj.onc.1209878. Citation on PubMed
- Neveling K, Endt D, Hoehn H, Schindler D. Genotype-phenotype correlations in Fanconi anemia. Mutat Res. 2009 Jul 31;668(1-2):73-91. doi: 10.1016/j.mrfmmm.2009.05.006. Epub 2009 May 21. Citation on PubMed
- Taniguchi T, D'Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006 Jun 1;107(11):4223-33. doi: 10.1182/blood-2005-10-4240. Epub 2006 Feb 21. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.