

Description
Encephalocraniocutaneous lipomatosis (ECCL) is a rare condition that primarily affects the brain, eyes, and skin of the head and face. Most of this condition's signs and symptoms are present from birth, and they vary widely among affected individuals.
A hallmark feature of ECCL is a noncancerous tumor under the scalp covered by a smooth, hairless patch of skin. This type of tumor, called a nevus psiloliparus, is made up of fatty tissue. Some people with ECCL also have noncancerous tumors under the skin elsewhere on the head or face. Many have small flaps of skin called skin tags on the eyelids and around the eyes. Hair loss (alopecia), thin or missing patches of skin on the scalp (dermal hypoplasia or aplasia), and changes in skin coloring (pigmentation) are also possible.
The most common eye abnormality in ECCL is a noncancerous growth called a choristoma. These growths can be present in one or both eyes and may affect vision.
About two-thirds of people with ECCL have noncancerous fatty tumors inside the brain or around the spinal cord. These tumors are called intracranial lipomas and intraspinal lipomas, respectively. Affected individuals also have an increased risk of developing a type of brain cancer called a glioma. The brain and spinal cord abnormalities associated with ECCL can cause seizures, abnormal tensing of the muscles, and intellectual disability ranging from mild to profound. However, about one-third of affected individuals have normal intelligence.
Other kinds of growths may also occur in people with ECCL, including noncancerous jaw tumors.
Frequency
ECCL is a rare disorder. Fewer than 60 cases have been reported in the medical literature.
Causes
ECCL can result from mutations in the FGFR1 gene, which provides instructions for making a protein called fibroblast growth factor receptor 1 (FGFR1). This receptor interacts with proteins called fibroblast growth factors (FGFs) to trigger signaling within cells. Signaling via the FGFR1 protein is involved in many critical processes, such as cell division and the regulation of cell growth and maturation. This signaling is important for the normal development and growth of several parts of the body, including the brain.
The FGFR1 gene mutations that cause ECCL arise randomly in one cell during the early stages of development before birth. As cells continue to grow and divide, some cells will have the mutation and others will not. This mixture of cells with and without a genetic mutation is known as mosaicism. In cells with an altered FGFR1 gene, the resulting FGFR1 protein is overactive, triggering abnormal signaling that affects cell growth and division. Researchers are studying how these changes in signaling lead to the growth of noncancerous tumors and the other features of ECCL.
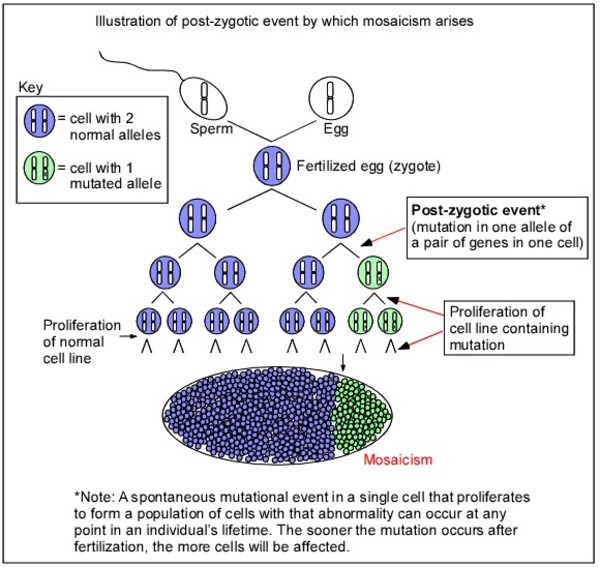
In some people with ECCL, no FGFR1 gene mutation has been identified, and the cause of the disease is unknown. Other genetic changes are under study as possible causes of this condition.
Inheritance
This condition is described as sporadic because it occurs in people without a history of the disorder in their family. Because ECCL results from mosaic gene mutations, which occur after conception, individuals do not inherit the condition from their parents.
Other Names for This Condition
- ECCL
- Fishman syndrome (formerly)
- Haberland syndrome (formerly)
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bennett JT, Tan TY, Alcantara D, Tetrault M, Timms AE, Jensen D, Collins S, Nowaczyk MJM, Lindhurst MJ, Christensen KM, Braddock SR, Brandling-Bennett H, Hennekam RCM, Chung B, Lehman A, Su J, Ng S, Amor DJ; University of Washington Center for Mendelian Genomics; Care4Rare Canada Consortium; Majewski J, Biesecker LG, Boycott KM, Dobyns WB, O'Driscoll M, Moog U, McDonell LM. Mosaic Activating Mutations in FGFR1 Cause Encephalocraniocutaneous Lipomatosis. Am J Hum Genet. 2016 Mar 3;98(3):579-587. doi: 10.1016/j.ajhg.2016.02.006. Citation on PubMed or Free article on PubMed Central
- Hunter AG. Oculocerebrocutaneous and encephalocraniocutaneous lipomatosis syndromes: blind men and an elephant or separate syndromes? Am J Med Genet A. 2006 Apr 1;140(7):709-26. doi: 10.1002/ajmg.a.31149. Citation on PubMed
- Moog U, Jones MC, Viskochil DH, Verloes A, Van Allen MI, Dobyns WB. Brain anomalies in encephalocraniocutaneous lipomatosis. Am J Med Genet A. 2007 Dec 15;143A(24):2963-72. doi: 10.1002/ajmg.a.32074. Citation on PubMed
- Moog U. Encephalocraniocutaneous lipomatosis. J Med Genet. 2009 Nov;46(11):721-9. doi: 10.1136/jmg.2009.066068. Epub 2009 Jul 1. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.