

Description
Crouzon syndrome with acanthosis nigricans is a disorder characterized by the premature joining of certain bones of the skull (craniosynostosis) during development and a skin condition called acanthosis nigricans.
The signs and symptoms of Crouzon syndrome with acanthosis nigricans overlap with those of a similar condition called Crouzon syndrome. Both conditions involve premature fusion of the skull bones, which affects the shape of the head and face. Other common features of both conditions include wide-set, bulging eyes due to shallow eye sockets; eyes that do not point in the same direction (strabismus); a small, beaked nose; and a flat or sunken appearance of the middle of the face (midface hypoplasia). Less common features that can occur in either disorder include an opening in the roof of the mouth (cleft palate), dental problems, or hearing loss. People with Crouzon syndrome or Crouzon syndrome with acanthosis nigricans usually have normal intelligence.
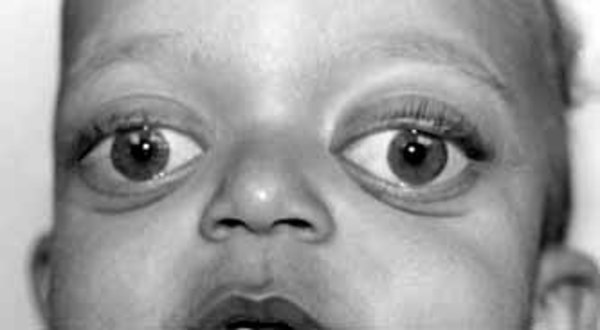
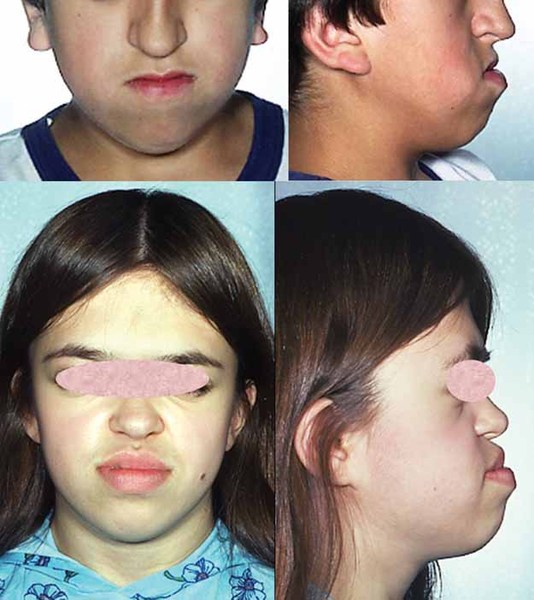

Crouzon syndrome with acanthosis nigricans is distinguished from Crouzon syndrome by several features, including skin abnormalities. Acanthosis nigricans is a skin condition characterized by thick, dark, velvety skin in body folds and creases, including the neck and underarms. People with Crouzon syndrome with acanthosis nigricans may also have other skin abnormalities; for example, scars in the thick, dark areas of skin are flat and pale. These scars are usually from surgical procedures that are commonly needed in affected individuals. Additionally, in some people with the condition, one or both nasal passages are narrowed (choanal stenosis) or completely blocked (choanal atresia), which can cause difficulty breathing. A buildup of fluid in the brain (hydrocephalus) can also occur. Nasal passage abnormalities and hydrocephalus are rare in Crouzon syndrome. Less common features of Crouzon syndrome with acanthosis nigricans include subtle changes in the bones of the spine (vertebrae), abnormalities of the finger bones, and noncancerous growths in the jaw called cementomas.
Frequency
Crouzon syndrome with acanthosis nigricans is rare; this condition occurs in about 1 person per million. For unknown reasons, it affects females more than twice as often as males.
Causes
A mutation in the FGFR3 gene causes Crouzon syndrome with acanthosis nigricans. This gene provides instructions for making a protein that is involved in the development and maintenance of bone and other tissues. The genetic change involved in this disorder causes the FGFR3 protein to be overly active, which disrupts the normal growth of skull bones and affects skin pigmentation. These changes lead to the features of Crouzon syndrome with acanthosis nigricans.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
In some cases, an affected person inherits the mutation from one affected parent. More commonly, this condition results from new (de novo) mutations in the gene. These cases occur in people with no history of the disorder in their family.


Other Names for This Condition
- CAN
- Crouzonodermoskeletal syndrome
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Arnaud-Lopez L, Fragoso R, Mantilla-Capacho J, Barros-Nunez P. Crouzon with acanthosis nigricans. Further delineation of the syndrome. Clin Genet. 2007 Nov;72(5):405-10. doi: 10.1111/j.1399-0004.2007.00884.x. Citation on PubMed
- Chen F, Degnin C, Laederich M, Horton WA, Hristova K. The A391E mutation enhances FGFR3 activation in the absence of ligand. Biochim Biophys Acta. 2011 Aug;1808(8):2045-50. doi: 10.1016/j.bbamem.2011.04.007. Epub 2011 Apr 22. Citation on PubMed or Free article on PubMed Central
- Chen F, Sarabipour S, Hristova K. Multiple consequences of a single amino acid pathogenic RTK mutation: the A391E mutation in FGFR3. PLoS One. 2013;8(2):e56521. doi: 10.1371/journal.pone.0056521. Epub 2013 Feb 20. Citation on PubMed or Free article on PubMed Central
- Cohen MM Jr. Let's call it "Crouzonodermoskeletal syndrome" so we won't be prisoners of our own conventional terminology. Am J Med Genet. 1999 May 7;84(1):74. No abstract available. Citation on PubMed
- Mir A, Wu T, Orlow SJ. Cutaneous features of Crouzon syndrome with acanthosis nigricans. JAMA Dermatol. 2013 Jun;149(6):737-41. doi: 10.1001/jamadermatol.2013.3019. Citation on PubMed
- Schweitzer DN, Graham JM Jr, Lachman RS, Jabs EW, Okajima K, Przylepa KA, Shanske A, Chen K, Neidich JA, Wilcox WR. Subtle radiographic findings of achondroplasia in patients with Crouzon syndrome with acanthosis nigricans due to an Ala391Glu substitution in FGFR3. Am J Med Genet. 2001 Jan 1;98(1):75-91. Citation on PubMed
- Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr Rev. 2000 Feb;21(1):23-39. doi: 10.1210/edrv.21.1.0387. Citation on PubMed
- Wenger T, Miller D, Evans K. FGFR Craniosynostosis Syndromes Overview. 1998 Oct 20 [updated 2020 Apr 30]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1455/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.