

Description
Alpha thalassemia is a blood disorder that reduces the production of hemoglobin. Hemoglobin is the protein in red blood cells that carries oxygen to cells throughout the body.
In people with the characteristic features of alpha thalassemia, a reduction in the amount of hemoglobin prevents enough oxygen from reaching the body's tissues. Affected individuals also have a shortage of red blood cells (anemia), which can cause pale skin, weakness, fatigue, and more serious complications.
Two types of alpha thalassemia can cause health problems. The more severe type is known as hemoglobin Bart hydrops fetalis syndrome, which is also called Hb Bart syndrome or alpha thalassemia major. The milder form is called HbH disease.
Hb Bart syndrome is characterized by hydrops fetalis, a condition in which excess fluid builds up in the body before birth. Additional signs and symptoms can include severe anemia, an enlarged liver and spleen (hepatosplenomegaly), heart defects, and abnormalities of the urinary system or genitalia. Without treatment, most babies with this condition are stillborn or die soon after birth because of these serious health problems. Hb Bart syndrome can also cause serious complications for women during pregnancy, including dangerously high blood pressure with swelling (preeclampsia), premature delivery, and abnormal bleeding.
HbH disease causes mild to moderate anemia, hepatosplenomegaly, and yellowing of the eyes and skin (jaundice). The features of HbH disease usually appear in early childhood, and affected individuals typically live into adulthood.
Frequency
Alpha thalassemia is a fairly common blood disorder worldwide. Thousands of infants with Hb Bart syndrome and HbH disease are born each year, particularly in Southeast Asia. Alpha thalassemia also occurs frequently in people from Mediterranean countries, Africa, the Middle East, India, and Central Asia.
Causes
Alpha thalassemia typically results from deletions involving the HBA1 and HBA2 genes. Less commonly, changes to the DNA sequence in or near these genes cause alpha thalassemia. Such changes are often referred to as nondeletion variants. Both the HBA1 and HBA2 genes provide instructions for making a protein called alpha-globin, which is a component (subunit) of hemoglobin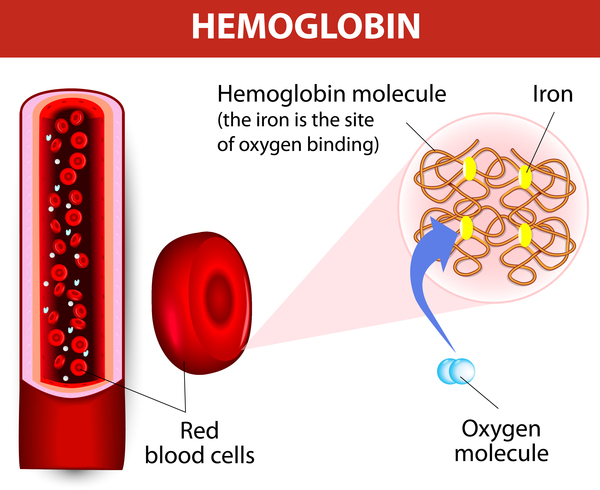 .
.
People have two copies of the HBA1 gene and two copies of the HBA2 gene in each cell. Each copy is called an allele. For each gene, one allele is inherited from a person's father, and the other is inherited from a person's mother. As a result, there are four alleles that produce alpha-globin. The different types of alpha thalassemia result from the loss or alteration of some or all of these alleles.
Deletions and nondeletion variants in one or more alleles reduce the amount of alpha-globin cells produce. Nondeletion variants tend to reduce alpha-globin more than deletions.
Hb Bart syndrome, the most severe form of alpha thalassemia, results from the loss or alteration of all four alpha-globin alleles. Such changes prevent the production of any normal alpha-globin.
HbH disease is usually caused by loss or alteration of three of the four alpha-globin alleles, which sharply reduces the amount of normal alpha-globin produced. Because nondeletion variants are usually more severe than deletions, nondeletion variants in two of the four alpha-globin alleles can result in HbH disease.
With a shortage of alpha-globin, cells make little or no normal hemoglobin. Instead, cells produce abnormal forms of hemoglobin called hemoglobin Bart (Hb Bart) or hemoglobin H (HbH). These abnormal hemoglobin molecules cannot effectively carry oxygen to the body's tissues. The substitution of Hb Bart or HbH for normal hemoglobin causes anemia and the other serious health problems associated with alpha thalassemia.
and the other serious health problems associated with alpha thalassemia.
Two additional forms of alpha thalassemia are related to a reduced amount of alpha-globin. Because cells still produce some normal hemoglobin, these forms tend to cause few or no health problems. A loss of two of the four alpha-globin alleles results in alpha thalassemia trait. People with alpha thalassemia trait may have unusually small, pale red blood cells and mild anemia. A loss of one alpha-globin allele is found in alpha thalassemia silent carriers. These individuals typically have no thalassemia-related signs or symptoms.
Nondeletion variants in one or two alleles cause a range of conditions, from alpha thalassemia silent carrier to HbH disease, depending on the allele involved.
Inheritance
The inheritance of alpha thalassemia is complex. Each person inherits two alpha-globin alleles from each parent. If both parents are missing at least one alpha-globin allele, their children are at risk of having Hb Bart syndrome, HbH disease, or alpha thalassemia trait. The precise risk depends on how many alleles are missing and which combination of the HBA1 and HBA2 genes is affected.
Other Names for This Condition
- Alpha-thalassemia
- Α-thalassemia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003 Feb 1;101(3):791-800. doi: 10.1182/blood-2002-07-1975. Epub 2002 Sep 12. No abstract available. Citation on PubMed
- Chui DH. Alpha-thalassemia: Hb H disease and Hb Barts hydrops fetalis. Ann N Y Acad Sci. 2005;1054:25-32. doi: 10.1196/annals.1345.004. Citation on PubMed
- Higgs DR, Weatherall DJ. The alpha thalassaemias. Cell Mol Life Sci. 2009 Apr;66(7):1154-62. doi: 10.1007/s00018-008-8529-9. Citation on PubMed
- Leung WC, Leung KY, Lau ET, Tang MH, Chan V. Alpha-thalassaemia. Semin Fetal Neonatal Med. 2008 Aug;13(4):215-22. doi: 10.1016/j.siny.2008.02.006. Epub 2008 Apr 10. Citation on PubMed
- Tamary H, Dgany O. Alpha-Thalassemia. 2005 Nov 1 [updated 2020 Oct 1]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1435/ Citation on PubMed
- Urbinati F, Madigan C, Malik P. Pathophysiology and therapy for haemoglobinopathies. Part II: thalassaemias. Expert Rev Mol Med. 2006 May 9;8(10):1-26. doi: 10.1017/S1462399406010805. Citation on PubMed
- Vichinsky EP. Alpha thalassemia major--new mutations, intrauterine management, and outcomes. Hematology Am Soc Hematol Educ Program. 2009:35-41. doi: 10.1182/asheducation-2009.1.35. Citation on PubMed
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704-12. Epub 2001 Oct 24. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.